| |
Role of Hepatitis B Viral Load and Basal Core Promoter Mutation in Hepatocellular Carcinoma in Hepatitis B Carriers
|
| |
| |
"....In the present cross-sectional, retrospective study.... In summary, the BCP T1762/A1764 mutation is the strongest viral factor associated with the development of HCC in hepatitis B carriers. Detection of the BCP T1762/A1764 mutation in HBV carriers may aid in the identification of chronic carriers at high risk for developing HCC, and they may be encouraged more aggressively to receive antiviral treatment and to undergo more active surveillance for HCC. In addition, HBV carriers with HBV DNA levels >105 copies/mL have an at least 2-fold excess risk of developing HCC, which provides useful information for setting up the goal of antiviral therapy. Finally, further studies regarding HBV-related hepatocarcinogenesis should focus on the structure and function of the X protein...."
The Journal of Infectious Diseases April 15, 2006;193:1258-1265
Chun-Jen Liu,1 Bing-Fang Chen,5 Pei-Jer Chen,1,2 Ming-Yang Lai,1,2 Wen-Ling Huang,1 Jia-Horng Kao,1,2,3,4 and Ding-Shinn Chen1,2
1Division of Gastroenterology, Department of Internal Medicine, 2Graduate Institute of Clinical Medicine, 3Hepatitis Research Center, and 4Department of Medical Research, National Taiwan University College of Medicine and National Taiwan University Hospital, and 5School of Medicine, Fu Jen Catholic University, Taipei, Taiwan
ABSTRACT
Background. Several hepatitis B viral factors correlate with the progression of chronic liver disease. However, the independent and interactive effects of each known viral factor on the development of hepatocellular carcinoma (HCC) remain largely unknown.
Methods. In a cross-sectional, retrospective, hospital-based setting, we comprehensively compared viral factors in 160 chronic hepatitis B virus (HBV) carriers and 200 patients with HCC, to clarify the independent and joint effect of each factor.
Results.
In univariate analysis, statistically significant odds ratios (ORs) were obtained for male sex (P < .001), advanced age (P < .001), HBV genotype C infection (P = .005), the precore A1896 mutation (P < .001), and the basal core promoter (BCP) T1762/A1764 mutation (P < .001).
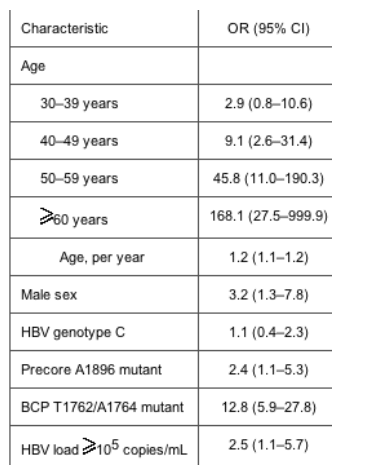
According to the results of multiple logistic-regression analysis, advanced age, male sex, the precore A1896 mutation, the BCP T1762/A1764 mutation, and an HBV load >105 copies/mL were independently associated with the development of HCC. Compared with patients with an HBV load <105 copies/mL (100,000) and the BCP A1762/G1764 wild-type strain, the adjusted OR of developing HCC was >30 in patients with an HBV load >105 copies/mL and the BCP T1762/A1764 mutant, irrespective of the presence of the precore A1896 mutation and viral genotype.
Conclusions. HBV load and the BCP T1762/A1764 mutation are important in hepatocarcinogenesis.
Joint effects of viral factors. We further combined various viral factors, to examine their joint effects on the development of HCC. Compared with an HBV load <105 copies/mL and the BCP A1762/G1764 wild-type strain, the adjusted OR of the development of HCC was 30 in patients with an HBV load >105 copies/mL (100,000 copies/ml) and the BCP T1762/A1764 mutation, irrespective of precore 1896 status (table 5).
Patients with an HBV load <105 copies/mL and infected with the precore A1896 and BCP T1762/A1764 mutants had a 21.8-fold (95% CI, 6.2-76.1-fold) increased risk of developing HCC, compared with patients infected with the precore G1896 and BCP A1762/G1764 wild-type strains (table 5). The effect of the precore A1896 mutation on the development of HCC was mainly found in patients infected with BCP T1762/A1764 mutants but only minimally in those infected with the BCP A1762/G1764 wild-type strain. On the other hand, considering patients with an HBV load >105 copies/mL, the major effect of the precore A1896 mutation on the development of HCC was found in patients with infected the BCP A1762/G1764 wild-type strain but not in those infected with the BCP T1762/A1764 mutant.
Table 4. Multivariate analysis of possible factors associated with the development of hepatocellular carcinoma.
Background
Hepatitis B virus (HBV) infection is a global health problem, and >350 million people in the world are chronic carriers of the virus [1]. The clinical manifestations of HBV infection include acute self-limiting infection or fulminant hepatic failure, the inactive carrier state, chronic hepatitis with progression to cirrhosis, and hepatocellular carcinoma (HCC) [2]. The pathogenesis of HBV infection is usually through interactions between the virus and host immune responses to HBV-encoded antigens [3]. Nevertheless, HCC occurs in a small proportion of hepatitis B surface antigen (HBsAg) carriers. It has been speculated that HBV-related hepatocarcinogenesis depends on several predisposing factors [4]. Apart from host factors, it is important to identify the relevant viral factors that likely are involved in the development of HCC.
At present, 8 HBV genotypes (A-H) have been identified [5-7]. Although the clinical significance of different HBV genotypes remains to be firmly settled, it has been shown that genotype C is associated with the development of HCC [8-11] and has a lower response rate to conventional interferon therapy than does genotype B [12, 13]. However, the molecular and virologic factors that contribute to these clinical and therapeutic differences among HBV genotypes need further examination.
Because of the spontaneous error rate of viral reverse transcriptase, the HBV genome evolves under the antiviral pressure of host immunity or specific therapy [14]. These HBV mutants could display an alteration of epitopes that are important in host immune recognition, enhanced virulence with the increased replication of HBV, resistance to antiviral therapies, or facilitated cell attachment/penetration [15]. Among these mutants, previous studies have revealed ample evidence that precore stop-codon mutations, as well as mutations in the basal core promoter (BCP), may be associated with severe and fulminant outcomes of HBV infection [14, 15]. The predominant mutation of the precore region involves a G⇒A change at nt 1896, which creates a premature stop codon at codon 28. Isolates with an A⇒T transversion at nt 1762, together with a G⇒A transition at nt 1764 (T1762/A1764) in the BCP (nt 1742-1849) are often present in hepatitis B carriers with chronic hepatitis, fulminant hepatitis, and HCC and less often in inactive carriers and immunosuppressed patients [15-18]. In addition, the BCP T1762/A1764 mutant has been reported to be associated with a poor response to interferon therapy in patients with chronic hepatitis B infection [19]. Several recent studies have also indicated that genotype C has a higher frequency of BCP T1762/A1764 mutations than does genotype B [12, 20]. Our previous study clearly demonstrated that HBV carriers with the BCP T1762/A1764 mutant have an increased risk of developing HCC [21]. In addition to viral genotype and mutants, recent studies have suggested that high HBV loads are also associated with the progression of chronic liver disease and an increased risk of developing HCC [22-29]. Beyond clinical outcomes, serum HBV loads have been associated with the response to antiviral treatments [30-32].
Several viral factors have seemed to influence the clinical outcomes of chronic HBV infection in previous studies, including ours [16-32]. Nevertheless, confounders may exist when only one or a few viral factors are analyzed each time. In the present study, we thus comprehensively investigated the independent and interactive effects of each known viral factor on the development of HCC in a hospital-based, HBV-infected population.
RESULTS
Demographic and Virologic Characteristics of Patients
Overall, patients with HCC were significantly older and more predominantly male than were chronic carriers. The prevalence of precore A1896 and BCP T1762/A1764 mutants and the distribution of HBV genotype and serum HBV load in both groups of patients are shown in table 1. The precore 1896 and BCP 1762/1764 mutations could not be determined in 2 patients, and mixed-genotype HBV infection was noted in 17 (3 in inactive carriers and 14 in patients with HCC).
Viral Factors Associated with the Development of HCC
When all the demographic and virologic features were compared in a univariate analysis between chronic carriers and patients with HCC, we found that statistically significant odds ratios (ORs) were obtained for male sex (P < .001), advanced age (P < .001), HBV genotype C (P = .005), the precore A1896 mutation (P < .001), and the BCP T1762/A1764 mutation (P < .001) (table 1).
Subgroup Analysis
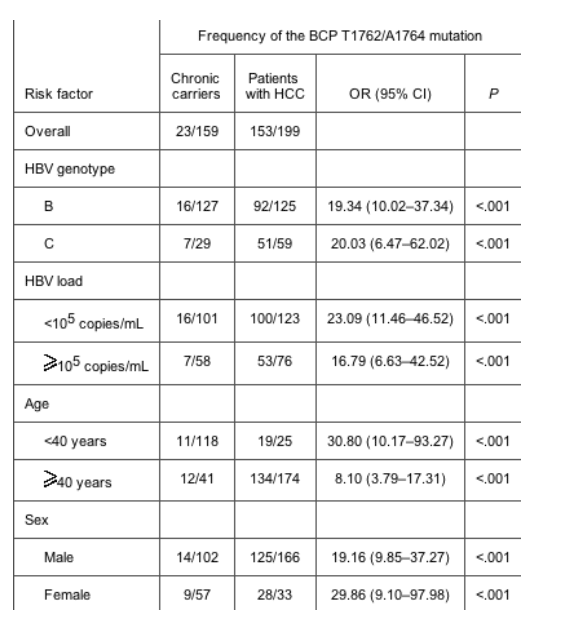
BCP mutation. The BCP T1762/A1764 mutation was the strongest factor associated with the development of HCC (table 1). The frequency of the BCP T1762/A1764 mutation increased from 14.4% in chronic carriers to 76.9% in patients with HCC (P < .001). Subgroup analysis revealed that the association existed irrespective of age, sex, HBV genotype, and HBV load (table 2). Nevertheless, when patients with HCC were compared with chronic carriers, the ORs for the frequency of the BCP T1762/A1764 mutation was particularly high in young patients, women, and those with a low HBV load (<105 copies/mL).
Table 2. The basal core promoter (BCP) T1762/A1764 mutation as a risk factor for the development of hepatocellular carcinoma (HCC) in subgroup analysis.
HBV load. Although the serum HBV loads were not significantly different between patients with HCC and chronic carriers in univariate analysis (P = .73), the distribution of serum HBV load varied with regard to the age of patients. In chronic carriers, the serum HBV load decreased gradually as age increased. In sharp contrast, the serum HBV load was significantly higher in older patients with HCC than in younger patients with HCC (log10 ± SD HBV DNA titer, 4.8 ± 1.4 vs. 4.2 ± 1.3; P < .05). In comparison with older chronic carriers (>40 years old), serum HBV loads were also significantly higher in older patients with HCC (log10 ± SD HBV DNA titer, 4.8 ± 1.4 vs. 3.6 ± 1.5; P < .05). By contrast, the serum HBV load was significantly lower in younger patients with HCC than in age-matched chronic carriers (log10 ± SD HBV DNA titer, 4.2 ± 1.3 vs. 5.3 ± 2.4; P < .05).
Genotype. Overall, when 88 patients infected with HBV genotype C were compared with 254 patients infected with HBV genotype B, genotype C was associated with a significantly higher incidence of the BCP T1762/A1764 mutation (65.9% vs. 42.9%; P < .001) (table 3). The distribution of serum HBV loads was similar between patients infected with HBV genotypes B and C. Subgroup analysis between patients with HCC who were infected with HBV genotype B and C revealed that the distribution of the precore A1896 mutation and an HBV load >105 copies/mL were also similar (80.8% vs. 88.0% [P = .65] and 63.5% vs. 52.5% [P = .16], respectively). However, patients infected with HBV genotype C remained marginally significantly associated with a higher proportion of the BCP T1762/A1764 mutation (86.4% vs. 73.6% [P = .051]) (table 3).
Cirrhosis. Cirrhosis, diagnosed either by histologic or ultrasonographic examination, was noted in none of chronic carriers and in 42.4% of patients with HCC. When patients with HCC and cirrhosis were compared with patients without cirrhosis, the distribution of age, sex, the precore A1896 mutation, the BCP T1762/A1764 mutation, and HBV load were comparable (data not shown). However, patients with cirrhosis tended to be infected with HBV genotype C and have an HBV load >105 copies/mL, compared with patients without cirrhosis (39.1% vs. 26.5% [P = .09] and 47.5% vs. 32.9% [P = .06], respectively).
DISCUSSION
In the present cross-sectional, retrospective study, the likelihood of developing HCC was independently associated with male sex, advanced age, increasing serum HBV loads, and the presence of the precore A1896 and BCP T1762/A1764 mutations. In addition, a joint effect between HBV load and the BCP T1762/A1764 mutation was observed. After adjustment for age and sex, patients infected with the BCP T1762/A1764 mutant who had an HBV load >105 copies/mL had a >30-fold increase in the risk of developing HCC, compared with patients infected with the BCP A1762/G1764 wild-type strain and a low HBV load, irrespective of viral genotype and precore 1896 status. This high relative risk indicates that both the BCP T1762/A1764 mutation and HBV load are important in better defining HBV carriers at high risk for progression to HCC.
HBV DNA consists of 4 overlapping genes that encode the viral envelope, nucleocapsid, polymerase, and X protein. Enhancer II and the core promoter are the regulatory sequences located at the downstream end of the X gene [36]. They control the transcription not only of the X gene but also of both precore mRNA and pregenomic RNA during the replication cycle of HBV [37]. At present, the most well defined BCP mutant has a dual change of T1762 and A1764 that diminishes the production of HBeAg and the resultant increased host immune response [15, 38]. It has been proposed that changes in the secondary structure of the pregenome that give rise to the T1762/A1764 mutation may increase viral replication [39]. This mutation can enhance the synthesis of core protein by 15-fold in an expression system [40] and create a binding site for hepatocyte nuclear factor-1 transcription factor, which may increase the transcription of pregenomic RNA [41]. Whether these observations are relevant to the enhanced virulence of the BCP T1762/A1764 HBV mutant remains to be established. After the thorough investigation of known viral factors possibly involved in the pathogenesis of HCC, we found that the BCP T1762/A1764 mutation was indeed the strongest independent predictor of the development of HCC [21]. Nevertheless, our findings did not support that the BCP T1762/A1764 mutation may increase HBV replication in patients with HCC (serum viral load for BCP T1762/A1764 wild-type vs. mutant, 9.7 X 103 vs. 2.1 X 104 copies/mL in the overall population [P = .12] and 9.1 X 104 vs. 2.5 X 104 copies/mL in patients with HCC [P = .13]). Furthermore, in multivariate analysis, the BCP T1762/A1764 mutation appeared to affect the risk of developing HCC independently of the serum HBV load. Thus, other mechanisms involved in BCP T1762/A1764 mutation-related hepatocarcinogenesis should be considered.
Alternatively, the X gene of the HBV genome encodes 2 proteins that have potent transactivation activities on viral as well as cellular genes [42]. This property makes the X gene a candidate for a role in the development of HCC in patients with chronic HBV infection. Several studies have indicated a direct interaction of X protein with p53, as well as with DNA repair factor, which suggests a direct link to cellular DNA repair, cell growth, and apoptosis [43]. Taken together, these lines of evidence suggest that the X protein may be important in hepatocarcinogenesis. Because its coding sequence overlaps regions of crucial importance for viral replication, such as enhancer II and the core promoter, mutations (including BCP T1762/A1764) in this region may therefore induce not only an alteration in HBV gene expression but also an amino acid change in the X protein [14, 44, 45]. In terms of the clinical situation, Takahashi et al. [46, 47] suggested that the frequency of the BCP T1762/A1764 mutant increases with disease progression in HBV genotype C-infected Japanese HBsAg carriers; they also indicated a high frequency (90%) of this mutant in patients with HCC. Similarly, a higher prevalence of the BCP T1762/A1764 mutant in black Africans with HCC than in asymptomatic carriers (66% vs. 11%; P < .001) was recently reported in South Africa, where HBV genotype E is prevalent [45]. In our patients, the BCP T1762/A1764 mutation was strongly associated with the development of HCC, irrespective of age, sex, viral genotype, and HBV load. In particular, compared with inactive carriers, the ORs for the frequency of the BCP T1762/A1764 mutation in patients with HCC were obviously higher in young patients, women, and those with a low HBV load. Whether BCP mutations and the resulting alteration in the amino acid sequences of the X protein play a role in hepatocarcinogenesis and in different subpopulations awaits further study.
Although the detailed mechanisms of hepatocarcinogenesis remain unclear, chronic HBV infection has been shown to cause HCC by inducing a long-term process of liver-cell necrosis, inflammation, and regeneration that is closely linked to the serum HBV load. Several lines of evidence support this speculation. First, high HBV loads and HBeAg positivity have been found to correlate with an increased risk of developing HBV-related HCC in prospective cohorts [28, 30]. Second, fluctuations in serum HBV load and HBeAg levels, which implies intermittent immune-mediated viral control and hepatocyte damage, have been associated with an increased HCC risk [25, 29]. Third, our previous studies demonstrated that profiles of serum ALT levels correlated well with the dynamics of serum HBV loads [35, 48]. Collectively, these studies have indicated that, during HBV DNA fluctuation, when liver-cell damage and the resulting turnover rate are accelerated, hepatocytes may be more vulnerable to the accumulation of genomic mutations or chromosomal instabilities. Actually, many genetic alterations have been observed during different stages of hepatocarcinogenesis [49-51]. From another standpoint, these facts support the idea that HBV DNA could serve as a useful marker for the progression of chronic liver disease and the development of HCC [22, 31]. Our multivariate analysis data consistently showed that the serum HBV load affected the risk of developing HCC. Subgroup analysis also revealed that the serum HBV load was significantly correlated with genotype B infection (OR, 2.6 [95% CI, 1.1-6.6]) and the presence of cirrhosis (P = .009). In particular, the joint effect of HBV load on the BCP T1762/A1764 mutation was most prominent in patients infected with the precore G1896 wild-type strain. The significance of the serum HBV load and its interaction with other viral factors in hepatocarcinogenesis and the progression of chronic liver disease need further examination.
In the present study, the association of HBV genotype C with the development of HCC was not evident, as we have reported elsewhere [21]. There are several possibilities that might explain the discordant results. First, because this was a retrospective, cross-sectional study, the effects of cohorting could not be excluded. Second, the study population was different from the previous one, so sampling bias should be considered. Third, because genotype C tends to have a higher proportion of BCP T1762/A1764 mutation [21], the effect of the mutation on the development of HCC might be strong enough to mask the effect of genotype C. In the future, large prospective studies should be conducted to clarify the role of each viral factor in the development of HCC. Furthermore, interactions among precore mutations, BCP mutations, and serum HBV loads should also be investigated.
The major limitation of our study was its retrospective design. Like most other series, this design cannot exclude the possibility of selection bias and examine other confounding factors. In addition, some important information\such as any family history of HCC\could not be obtained from each subject. However, a previous case-control study from Taiwan already suggested that first-degree relatives of patients with HCC are more likely to develop HCC (age- and sex-adjusted OR, 2.57 [95% CI, 2.03-3.25]) than are first-degree relatives of subjects without HCC [52]. Thus, the first-degree relatives of patients with HBV-related HCC appear to be at an increased risk of developing HCC. Although this association was not explored, our study was unique in the collection of patients with detailed clinical, histologic, and virologic data, which allowed us to assess the interactions among viral factors and the risk of developing HCC.
In summary, the BCP T1762/A1764 mutation is the strongest viral factor associated with the development of HCC in hepatitis B carriers. Detection of the BCP T1762/A1764 mutation in HBV carriers may aid in the identification of chronic carriers at high risk for developing HCC, and they may be encouraged more aggressively to receive antiviral treatment and to undergo more active surveillance for HCC. In addition, HBV carriers with HBV DNA levels >105 copies/mL have an at least 2-fold excess risk of developing HCC, which provides useful information for setting up the goal of antiviral therapy. Finally, further studies regarding HBV-related hepatocarcinogenesis should focus on the structure and function of the X protein.
PATIENTS, MATERIALS, AND METHODS
Patients. Three hundred sixty genotype B- or C-infected chronic HBV carriers have been treated at the gastroenterologic clinics of the National Taiwan University Hospital since 1990. They included 160 chronic HBV carriers (103 men and 57 women; 128 infected with genotype B, 29 with genotype C, and 3 with mixed genotype B and C; mean age, 32.5 ± 10.0 years) with persistently normal or intermittently mildly elevated serum alanine aminotransferase (ALT) levels for at least 3 years and 200 HBsAg-positive patients with histologically verified HCC (167 men and 33 women; 126 infected with genotype B, 59 with genotype C, 14 with mixed genotype B and C, and 1 with unclassified genotype; mean age, 54.0 ± 12.3 years) (table 1). Of the patients with HCC, 26 were <40 years old, and the remaining 174 were >40 years old. None of the clinical and virologic data of patients with HCC and carriers without HCC have been described in our previous reports [8, 21]. None of them had coinfection with hepatitis C or D virus.
Serum samples from each subject were stored at -70C until use. For patients with histologically verified HCC, serum samples were collected at the time when liver biopsy or surgical resection for HCC was performed.
Serologic markers. HBsAg, hepatitis B e antigen (HBeAg), antibodies against hepatitis C virus, and antibodies against hepatitis D virus were tested with commercial kits (Ausria II, IMx HBe 2.0, HCV EIA II, and Anti-Delta; Abbott Laboratories).
Quantification of HBV DNA. We developed a 1-tube assay for the quantification of HBV DNA, as described elsewhere [33]. The HBV loads obtained from this quantification assay showed a satisfactory consistency with Roche Amplicor and NGI SuperQuant assays (Pearson's > 0.99) in the linear range from 1 X 102 to 1 X 1011 copies/mL. The detection limit of our 1-tube assay for HBV DNA is 1 X 102 copies/mL.
Strict precautions were taken to avoid possible contamination. In addition, reagents were stored in small aliquots. All pipette tips and Eppendorf tubes were disposable, and pipette tips were filtered. Only data that revealed no false-positive results in the negative controls and that were reproducible were used.
Genotyping of HBV. HBV genotype was determined by using the line-probe assay (INNO-LiPA HBV genotyping assay; Innogenetics). In brief, HBV DNA was first extracted and amplification was performed by use of polymerase chain reaction (PCR) assay using primers in the HBV S gene region, as described elsewhere [34]. Samples with detectable HBV DNA after nested PCR were tested by the INNO-LiPA HBV genotyping assay in accordance with the manufacturer's instructions.
Determination of precore nt 1896 and BCP dinucleotide 1762/1764 mutations. The presence of the precore nt 1896 and BCP dinucleotide 1762/1764 mutations was also determined using the line-probe assay (INNO-LiPA HBV PreCore assay; Innogenetics). Except for the primers and reaction strips, the procedure was similar to that for HBV genotyping [12, 16]. The probes were designed to determine the nucleotide sequences at position 1896 in the precore region (G vs. A) and positions 1762 (A vs. T) and 1764 (G vs. A and G vs. T) in the BCP region. Multiple probes were applied on the strip for each motif, taking into account the extensive variability surrounding the specific nucleotide positions assessed by the assay.
Ethical considerations. The study was performed in accordance with the principles of the Declaration of Helsinki. The study was approved by the Ethical Committee of National Taiwan University Hospital, and serum samples were collected after informed consent was obtained from patients.
Statistical analysis. Continuous data are expressed as mean ± SD, and the categorical data are expressed as number (percentage). Differences in demographic and clinicopathologic features between inactive carriers and patients with HCC were evaluated by Pearson's 2 test, Fisher's exact test, and Student's t test, where appropriate. Logistic-regression analysis was used to assess the influence of each viral factor on the risk of developing HCC. P < .05 was considered to be statistically significant.
|
|
| |
| |
|
|
|