| |
Maintenance PegIntron Study in HCV/HIV Coinfected Patients
|
| |
| |
Reported by Jules Levin
"A Randomized, Open-label, Multi-center, Phase 3, 2-arm Study Evaluating the Efficacy and Safety of Peg interferon Alfa-2b Low-dose Maintenance Monotherapy Versus Standard Supportive Care in Patients With Cirrhotic Hepatitis C Co-infected With Human Immunodeficiency Virus - The ENDURE Study"
Principal Investigators:
MASSIMO PUOTI, MD University of Brescia, Italy)
MARK SULKOWSKI, MD (Johns Hopkins Medical School)
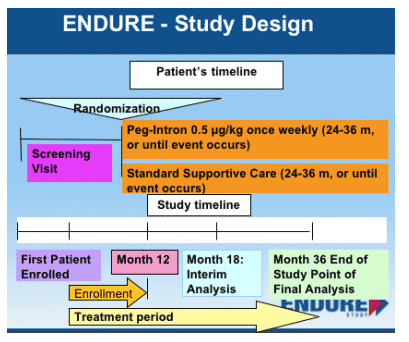
The initiation of the ENDURE Study was announced publicly by a Press Release from Schering Plough during the Intl Coinfection Workshop in Amsterdam (Jan 14, 2005). At the Workshop Massimo Puoti conducted a symposium and presented extensive information about the ENDURE Study including - Study Rationale & Objectives, Inclusion/Exclusion, Study Design, and Study Monitoring Boards. Puoti emphasized that although the study is 36 months long, the plan is to perform an interim analysis at 18 months. If ENDURE is able to enroll ASAP this would allow for the possibility of providing data late next year, allowing for immediate changes of guidelines relating to the use of maintenance therapy for patients.
STUDY POPULATION
Subjects: 448 adult subjects with chronic hepatitis C virus co-infected with HIV. Subjects will be compensated cirrhotics as defined by the Child-Pugh score of 8 or less.
STUDY RATIONALE
PegIntron long term maintenance improves the survival rate in NR subjects infected with HCV.
PegIntron may reduce the occurrence of death, decompensated liver disease, liver transplants and hepatocellular carcinoma (HCC) in subjects with HIV-HCV co-infection who did not respond to prior treatment or are not suitable for full dose treatment.
PRIMARY STUDY OBJECTIVE
To compare at End of Study the efficacy of:
PEG-IFN alfa-2b 0.5 mg/kg once weekly (QW) monotherapy versus standard supportive care for up to 36 months in subjects with cirrhotic hepatitis C coinfected with HIV, using the combined endpoint of death, liver decompensation, liver transplant, or hepatocellular carcinoma.
STATISTICAL
Calculated sample size
Log rank test, 2-stage group sequential analysis with Pocock alpha spending to maintain overall 0.05 significance level
Total of 448 patients will achieve over 80% power
Primary Hypothesis:
Null Hypothesis
Time to clinical event distributions are the same between the two treatment groups
Interim Analysis
35 clinical events AND >/= 1.5 years from first subject (Assume 1 year recruitment)
Time to clinical event is different if p-values
Final Analysis
Time to clinical event is different if p-values
Secondary Objective(s):
To compare the two treatment groups at the EOS (end of study):
The times to each of the following 5 clinical end points
- Overall mortality
- Liver-related mortality
- Liver decompensation
- Liver transplants
- Hepatocellular carcinoma (HCC)
The Incidence of:
- Overall mortality
- Liver-related mortality
- Liver decompensation
- Liver transplants
- Hepatocellular carcinoma (HCC)
Additional Secondary Objectives to be compared in the two treatment groups:
The incidence of total serious adverse events (SAEs) in the two groups.
To assess the effect of PEG-IFN low dose maintenance treatment on HIV-1 infection progression as determined by:
- changes in CD4 cell count levels,
- changes in HIV RNA titers,
- HIV related infections
- ART regimen changes.
Changes in serum ALT and HCV RNA between the two groups.
Changes in Fibro Scan and APRI during the study period between the two groups.
Changes in hepatic venous pressure gradient (HVPG) levels in a subgroup of subjects from the two treatment groups with available data.
Changes in proliferative histological indices and fibrotic scores in a subgroup of paired liver biopsies at the beginning and EOS. Liver biopsies will be obtained from the two treatment groups.
BASELINE STRATIFICATION
Subjects will be stratified at randomization according to:
- CD4 count (<200 vs. ≥200) and
- portal hypertension (defined clinically, existing vs. non-existing)
INCLUSIONS
Display the willingness and ability to participate in the study by demonstrating full comprehension of and agreement to comply with all study procedures by signing the written informed consent.
>/=18 years but < 70 years, of either sex or any race.
Detectable plasma HCV RNA, as measured by the Taqman (all genotypes of HCV are permitted). Historical PCR and genotype are acceptable for study entry and will be confirmed at screening through the central lab.
Cirrhosis of the liver, confirmed by histopathologic findings, within the last three years.
Compensated liver disease measured by the Child-Pugh clinical classification with the hepatic encephalopathy measure equal to 1 and the total score less than or equal to 8.
No evidence of HCC on either abdominal ultrasound, CT scan, or MRI scan, and a serum AFP <100 ng/mL within two months of randomization/study enrollment.
Varices results via endoscopy within the last six months or at time of screening.
Serologic evidence of HIV-1 infection by HIV-1 antibody or detection of HIV-1 RNA. (Historical HIV RNA is acceptable for study entry and will be confirmed at screening through a central lab).
CD4 cell count >100 /mL, regardless of HIV-1 RNA load.
Platelet number of at least 50000 mm3.
Neutrophil count of at least 750 mm3.
Hemoglobin of >9.0 mg%.
Any serum ALT/AST liver enzyme level.
Serum thyroid stimulating hormone levels within normal limits, regardless of treatment with L thyroxin.
HbA1c<8.5%, to demonstrate controlled diabetes, if applicable.
Written clearance from an ophthalmologist must be presented for subjects with a history of hypertension or diabetes prior to treatment start.
Creatinine clearance >50 mL/min, as assessed by the indirect calculation method (Appendix 8).
Demonstrate stable status of HIV-1 infection, in the opinion of the principal investigator, (e.g., subjects who are expected to progress in the first 3 months of the study would not be appropriate for enrollment).
On stable antiretroviral therapy (HAART) for at least 6 weeks prior to baseline, with the expectation of their HAART regimen (drugs and dosage) remaining unaltered for the first 8 weeks of the study.
OR
Willing to delay initiation of HAART therapy for at least 6 weeks (for subjects who have not been on HAART for at least 8 weeks prior to randomization).
"Structured treatment interruptions" will be permitted during the study.
Counseled in the appropriate use of birth control while in this study, as confirmed by the principal investigator or a sub-investigator.
Female subjects of childbearing potential (includes women who are less than two years post-menopausal and women who may become sexually active during the study) must agree to use a medically acceptable barrier method of contraception (or be surgically sterilized prior to screening) while receiving protocol-specified medication and for one month after stopping the medication.
Acceptable methods of contraception include condoms (male and female) with a spermicidal agent, diaphragm or cervical cap with spermicide, medically prescribed IUD, oral or injectable hormonal contraceptive, and surgical sterilization (e.g., hysterectomy or tubal ligation).
Male subjects who are sexually active must be using an acceptable method of contraception (vasectomy, condoms with spermicide). Contraception must be used during the treatment period and for one month after the completion of therapy, including condom use by male subjects with pregnant partners.
Negative serum pregnancy test (for women of childbearing potential) documented within the 24-hour period prior to the first dose of study drug.
Confirmation by the principal investigator or a sub-investigator that sexually active male/female subjects are practicing a method of contraception considered acceptable. Contraception must be used during the treatment period and for 1 month after the completion of therapy, including condom use by male subjects with pregnant partners. For subjects who take possible teratogenic HAART regimen (e.g., efavirenz) additional counseling should be given in regard to contraception.
Free of any clinically significant disease (other than HCV and HIV) that would interfere with study evaluations.
EXCLUSIONS
Any subject will be excluded from entry into the study if they have ANY of the following criteria:
Female who is pregnant, intends to become pregnant during the study or within two months after study completion, or is nursing. Male subjects whose partner wants to become pregnant.
Using silymarin.
Positive test at screening for anti-HAV IgM Ab, HBsAg, anti-HBc IgM Ab, or HBeAg.
Any cause of liver disease other than chronic hepatitis C, including-but not limited to:
Hemochromatosis
Alpha-1 antitrypsin deficiency
Wilson's disease
Autoimmune hepatitis
Alcoholic liver disease
Non-alcoholic steatohepatitis (NASH)
Drug-related liver disease
Suspected or having hypersensitivity to interferon.
History of liver decompensation status or other evidence of bleeding from esophageal varices, signs of current bleeding, significant ascites, hepatic encephalopathy, jaundice or other conditions consistent with decompensated liver disease.
Present with a lesion suspicious for hepatic malignancy (HCC or metastasis/metastases) on the screening imaging.
Any active malignant disease, suspicion, or history of malignant disease within 5 years prior of study enrollment (except for adequately treated basal cell carcinoma).
Known coagulation (e.g., hemophilia) or hemoglobin (e.g., thalassemia) diseases.
Organ transplant, except corneal or hair transplant.
Any known preexisting medical condition that, in the investigator's opinion, could interfere with the subject's participation in and completion of the study, such as:
-Preexisting psychiatric condition, especially moderate to severe depression, or a history of severe psychiatric disorder, such as psychosis, suicidal ideation, or suicide attempts. Severe depression includes the following:
- Hospitalization for depression
- Electroconvulsive therapy for depression, or
- Depression causing a prolonged absence from work or significantly altering
daily functions.
Subjects with mild depression may be considered for entry into the study provided that a pre-treatment assessment demonstrates that the subject's emotional status is clinically stable (either on or off drugs), in which case a management plan must be formulated for the subject; this management plan will become a part of the subject 's medical record.
Craniocerebral trauma which is not a concussion, or active seizure disorders requiring medication
Clinically significant ECG abnormalities and/or cardiovascular dysfunction within six previous months (e.g., angina, congestive heart failure, recent myocardial infarction, or significant arrhythmia)
Chronic lung disease (e.g., chronic obstructive lung disease)
Poorly controlled diabetes mellitus
Immune-mediated disease (e.g., inflammatory bowel disease [Crohn's disease, ulcerative colitis], idiopathic thrombocytopenic purpura, systemic lupus erythematosus, autoimmune hemolytic anemia, scleroderma, severe psoriasis)
Clinical gout
Active HIV-related opportunistic infection and/or malignancy requiring systemic therapy.
Evidence of known severe retinopathy (e.g., CMV retinitis, macular degeneration).
Subjects that in the investigator's opinion are non-reliable for the study conduct or therapy drug administration. These subjects may be - but not limited to - illicit drug/alcohol users whether on substitution therapy or not.
Subject has not observed the designated washout periods for any of the prohibited medications outlined in Section 7.2.
Participating in any other Hepatitis C clinical study. Subjects who chose to participate in other HIV investigational drugs study may do this if the other protocol allows.
Subject is part of the staff or a family member of the staff personnel directly involved with this study.
Any other condition that in the opinion of the investigator will make the subject unsuitable for enrollment or will interfere with the subject participating in or completing the study.
STUDY MONITORING BOARDS
Clinical Events Judification Committee
Liver decompensation
- Determine criteria
- Interim and Final Analysis
Data Safety Monitoring Board
- SAEs
- Discontinuation rates
- Laboratory results
|
|
| |
| |
|
|
|