| |
New HCV Drugs Discovery
|
| |
| |
The face of future hepatitis C antiviral drug development: Recent biological and virologic advances and their translation to drug development and clinical practice
Journal of Hepatology
Volume 44, Issue 2, Pages 411- 421 (February 2006)
John G. McHutchisona, Ralf Bartenschlagerb, Keyur Patela, Jean- Michel Pawlotskyc
a Division of Gastroenterology, Duke Clinical Research Institute, Duke University Medical Centre, 2400 Pratt Street, Room 0311, Terrace Level, Durham, NC 27707, USA
b Department for Molecular Virology, University Heidelberg, Heidelberg, Germany
c Department of Virology & INSERM U635, Hopital Henri Mondor, Universite Paris XII, Creteil, France
Conclusion
There is good reason for renewed optimism regarding future treatment options for HCV infected patients, and as outlined above, there are many newer agents currently in clinical development programs.
Advances in the development of in vitro cell culture systems should lead to a greatly improved understanding of host- viral interaction during the early phases of HCV infection. This may provide opportunities to develop novel therapeutic approaches that disrupt the HCV replication pathways prior to polyprotein synthesis. These and other future technological advances will allow a more expansive search for new molecular targets that may translate into new drugs. Thus, the goals of future therapies should be to provide better clinical outcomes by using multiple drugs that act through different mechanisms of action, while at the same time preventing the development of clinically significant resistance. Despite these very positive steps forward in identifying many new targets, one major remaining hurdle will be to transform compounds effective in cell- based systems into bio- available (i.e. with adequate levels of circulating free or active drug and adequate intracellular concentrations), safe and effective drugs.
Further progress in our understanding of the pathogenesis of HCV infection and identification of potential therapeutic targets are inherently dependent on continued development of cell culture systems and small animal models of infection. This should lead to a broader repertoire of therapeutic options for the clinician that is likely to be faced with an ever increasing proportion of HCV infected patients over the next decade.
Newer compounds should ideally provide effective, more tolerable therapy to a greater proportion of patients with this disease. We look forward to the laboratory/basic science development of future agents that will hopefully translate into greater efficacy in clinical trials, and then subsequently be translated into clinical practice, where the great majority of infected patients seek management and treatment.
Article Outline
- 1. Introduction
- 2. New systems to aid drug discovery
- 2.1. The replicon system
- 2.2. The pseudoparticle system
- 2.3. The infectious virus system
- 3. Advances in virology
- 3.1. Early steps of infection
- 3.2. Polyprotein synthesis
- 3.3. Post- translational processing
- 3.4. HCV replication
- 3.5. Virus assembly and release
- 4. Recent clinical hepatitis C drug developments
- 4.1. HCV specific protease inhibitors
- 4.2. Polymerase inhibitors
- 4.3. Immune modulators
- 4.3.1. Interferons
- 4.3.2. Toll- like receptor agonists
- 4.3.3. Ribavirin analogues
- 4.4. RNA- based therapy
- 4.5. Vaccines
- 5. Conclusion
1. Introduction
Worldwide, approximately 170 million people are infected with hepatitis C virus (HCV) [1], yet current therapies are only effective in approximately 50% of infected patients [2,3]. Unfortunately, such therapies are costly, prolonged, associated with significant side effects, and not suitable for many patients.
Important recent technical advances in cell culture systems and replication assays have led to discoveries related to the mechanisms of HCV infection and potential antiviral targets. Many novel approaches to HCV infection are now currently being evaluated. In this article we describe recent noteworthy advances in, in vitro assays, HCV specific virology, and the clinical development of potential new therapies.
2. New systems to aid drug discovery
While the introduction of pegylated interferon (pegIFN)- alfa and ribavirin combination therapy has led to an increase in rates of sustained viral response in the last few years, the treatment has several disadvantages. Most notable are the severe side effects and the limited efficacy against genotypes 1 and 4 viruses. Since the first molecular cloning of the HCV genome in 1988, prime targets for antiviral therapy have been defined, but progress in development of selective therapy has been slowed by the lack of cell culture systems that support efficient propagation of HCV in the laboratory. This hurdle has gradually been overcome in the past few years, starting with the development of the replicon system in 1999 [4].
2.1. The replicon system
The replicon system is based on the autonomous replication of genetically engineered HCV 'mini- genomes', in which the region encoding core to NS2 is replaced by a selectable marker (most often neo) and an internal ribosome entry site that mediates translation of the HCV replicase (NS3- 5B) (Fig. 1). Upon transfection into cells of the human hepatoma cell line Huh- 7 and G418- selection, cell clones that carry high amounts of viral RNA and proteins can be established. The success of this system as compared with infection- based cell assays with HCV derived from patient sera is due to the coupling of HCV RNA replication to host cell survival (reviewed in [5]). Transfected cells are G418 resistant only when replicon RNA levels are sufficiently high. The selection pressure leads to the accumulation of mutations that enhance RNA replication and to the selection for Huh- 7 cells that support RNA replication to a very high level. Cell culture adaptive mutations and a favorable host cell environment allow replication of HCV RNAs to an unprecedented level.
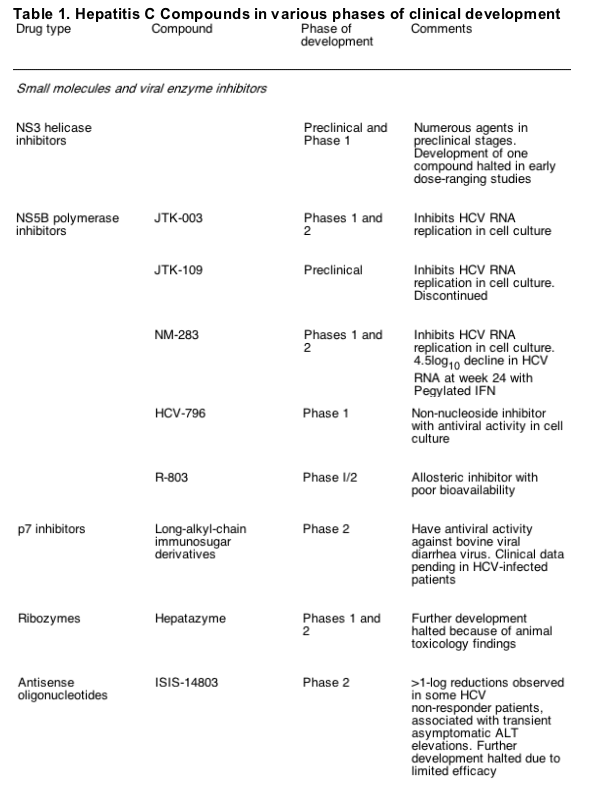
Fig. 1. Cell- based assays for the study of hepatitis C virus (HCV) replication in cell culture. (A) Structure of a subgenomic HCV replicon carrying a selectable marker (neo) that confers G418- resistance. Translation of neo is mediated by the HCV IRES, whereas NS3 to NS5B are translated under control of the encephalomyocarditis virus IRES. (B) Schematic of the structure of an HCVpp. It is composed of the envelope into which E1- E2 glycoprotein complexes are embedded, the HIV nucleocapsid (CA), and two copies of an HIV vector RNA carrying a reporter gene such as green fluorescent protein. (C) Generation of infectious HCV in cell culture. Huh- 7 cells are transfected with genomic HCV RNA depicted in the top (the authentic JFH- 1 genome or a JFH1- chimera in which the region from core up to the N- terminus of NS2 [indicated by shading] stems from another HCV isolate). Four days later, culture supernatant is harvested, used to inoculate naive Huh- 7 cells, and after 2 further days cells are analyzed for infection by using a NS3- specific immunofluorescence assay (lower panel). Infection was performed directly with supernatant (left panel) or with supernatant containing either a CD81- specific antibody or a control antibody (right and middle panel, respectively). Note the strong reduction of the number of infected cells when infections are performed in the presence of the CD81- specific antibody.
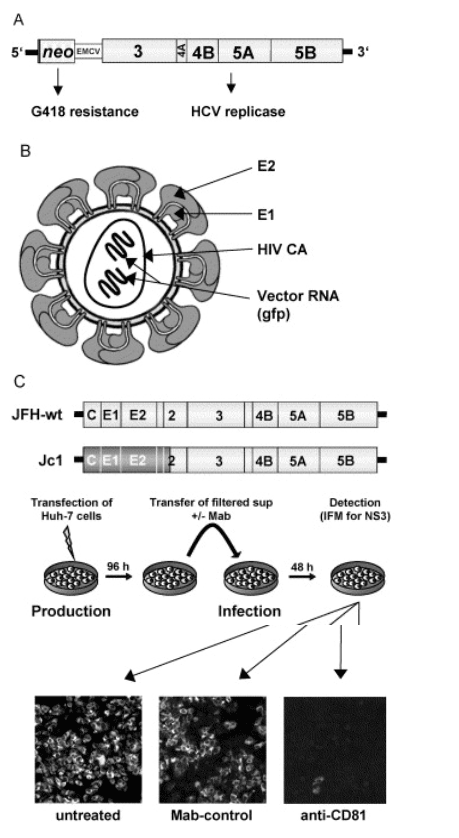
With these improvements, genomic replicons as well as replicons carrying stably integrated reporter genes have been constructed (reviewed in [6]). Replicons with reporter genes allow rapid and easy drug screening and can be adapted to 384- well plate assays. Moreover, replicons have been instrumental in evaluating compounds identified by rational drug design or screening assays with recombinant enzymes. However, HCV RNA replication occurs in membrane- associated replication complexes composed of viral and cellular proteins. Therefore, enzyme binding sites that are accessible in an isolated protein may be sterically blocked or masked in the replication complex (for an example, see [7]). In the last few years, replicons have been used to select for resistance against selective compounds targeting the NS3 protease and the NS5B RNA- dependent RNA polymerase (RdRp). Resistance mutations can arise against most, if not all, compounds targeting these enzymes. In several cases, these primary resistance mutations reduce replicon fitness [8], but it is likely that after prolonged passaging of replicon- harboring cells, the reduction in fitness would be compensated by secondary mutations that at least partially restore replication fitness. Whether and to what extent this will happen in vivo remains to be seen.
2.2. The pseudoparticle system
The second important model system for studying HCV replication in cell culture is the pseudoparticle (HCVpp) system [9,10]. It is based on the co- expression of the HCV envelope glycoproteins E1 and E2 together with a retroviral gag protein and a retroviral vector into which a reporter gene has been inserted. In transfected cells, the gag proteins that are derived either from the murine leukemia virus (MLV) or the human immunodeficiency virus (HIV) assemble into capsids that carry two copies of the vector RNA. The capsids are surrounded by a lipid envelope that carries native E1/E2 complexes (Fig. 1). Upon infection of target cells, the retroviral vector is liberated into the target cell, integrated into the host genome, and the reporter gene is expressed. The level of reporter gene expression is therefore a marker for the efficiency of infection governed by the HCV envelope glycoproteins. By using this system, the early steps of the HCV replication cycle (binding to and entry into the host cell) have been studied. Several molecules have been postulated to be involved in HCV entry (reviewed in [5]), however, none of these cell surface molecules are sufficient to mediate uptake, suggesting that HCV infection requires a receptor complex.
The HCVpp system has also been instrumental in determining the humoral immune response induced upon HCV infection in humans and chimpanzees. Neutralizing and cross- neutralizing antibodies are generated during natural infection, and the extent of the humoral immune response appears to correlate with viral clearance [11- 15].
2.3. The infectious virus system
Very recently, the efficient production of infectious HCV has been achieved [16- 18]. The development of this system began with the discovery of a genotype 2a isolate from a Japanese patient with fulminant hepatitis C. This isolate, designated JFH- 1, is able to replicate to very high levels in Huh- 7 cells in the absence of cell culture adaptive mutations [19]. The full length JFH- 1 genome was cloned, and a chimeric genome was constructed in which the region from core to the end of NS2 of another genotype 2a isolate was combined with the JFH- 1 replicase (NS3- 5B) and its non- translated regions. Upon transfection of Huh- 7 cells with both genomes, infectious HCV particles were secreted in a manner dependent upon envelope glycoproteins. Infectivity of these particles was demonstrated by (a) inoculation of naive Huh- 7 cells, (b) passage of the virus via infection of naive cells, and (c) inhibition of infection by E2- or CD81- specific antibodies, by the large extracellular loop of CD81, and by immunoglobulins from chronically infected hepatitis C patients. Moreover, virus particles generated from the authentic JFH- 1 full- length genome were infectious in a chimpanzee, confirming the authenticity of HCV grown in cell culture [17]. This virus has a heterogenous density that may be due to the association of the particles with host cell components such as lipoproteins. Immuno- EM studies have revealed spherical particles with an average diameter of about 55nm and an inner electron dense structure with a diameter of about 30nm that presumably represents the nucleocapsid.
Unfortunately, only the JFH- 1 replicase supports high- level HCV replication in the infectious HCV cell culture system. Numerous HCV isolates with proven infectivity do not or only very poorly replicate in tissue culture unless they acquire adaptive mutations (reviewed in [5]). In the case of the Con1 HCV isolate, from which the first HCV replicon was derived, introduction of cell culture adaptive mutations reduces or completely blocks production of virus particles (T. Pietschmann and R.B., unpublished). One possible mechanism to overcome this limitation is to generate chimeric genomes in which the prime drug targets of JFH- 1 are replaced by those of other isolates. Such chimeras would allow antiviral drugs to be evaluated in an infection- based assay against targets derived from different HCV isolates and genotypes. While it is unclear whether this approach will work with these replacements, the region encoding core to NS2 of JFH- 1 can be replaced by genes from several other isolates and genotypes (16; T.P. et al., unpublished) (Fig. 1). In addition, infectious HCV can be adapted to Huh- 7 cells with this adapted variant yielding much higher titers of infectious virus progeny as compared with the parental JFH- 1 (A. Kaul, T.P. and R.B., unpublished).
Another important extension of the infectious virus system is the construction of HCV genomes that stably express a reporter gene. For instance, a bicistronic JFH- 1 variant was generated that expresses the firefly luciferase gene [17]. Upon infection of host cells, replication can be measured with high sensitivity and accuracy by using luciferase assays. This approach is not limited to luciferase: variants that express other markers such as green fluorescent protein also yield infectious virus (T.P. et al., unpublished). These systems will be very useful for characterizing the early steps of HCV infection, and identifying neutralizing antibodies [17], novel drug targets, and evaluating existing compounds for their impact on virus production. Finally, the infectious virus system should simplify the selection for drug resistance, because it avoids the cumbersome selection of replicon cells with multiple concentrations of G418 (in case of neo replicons) and HCV- specific drug.
3. Advances in virology
The goal of anti- HCV therapy is to disrupt the equilibrium of HCV kinetics and clear all infected cells from the patient's body. At the chronic stage of infection, infected hepatocytes continuously produce large amounts of HCV virions. The vast majority of newly produced virions are released into the peripheral circulation where they are continuously degraded in a virtual degradation compartment by unknown mechanisms [20]. The steady- state viral kinetics of chronic hepatitis C are characterized by an equilibrium between hepatocyte infection and infected cell death, ensuring a constant size of the pool of infected hepatocytes, and an equilibrium between the release of newly produced HCV virions into the peripheral circulation and their degradation, ensuring stable viral load [20]. Anti- HCV therapy may act by reducing virion production (antiviral drugs) or by promoting the clearance of infected cells (essentially immune therapies) [21].
The HCV lifecycle has been poorly studied, principally because of the lack, until very recently, of a virion productive in vitro cell culture system as discussed above. Most of what is believed to occur has been deduced from studies with other members of the Flaviviridae family. Virtually any step of the HCV lifecyle represents a potential target for specific HCV inhibitor drugs.
3.1. Early steps of infection
Several cell surface molecules have been proposed to mediate HCV binding and internalization, including the CD81 molecule [22]; the scavenger receptor B type I (SR- BI) [23]; the dendritic cell- specific intercellular adhesion molecule- 3- grabbing non- integrin (DC- SIGN or CD209) and the liver/lymph node- specific intercellular adhesion molecule- 3 (ICAM- 3)- grabbing integrin (L- SIGN or CD209L) [24,25]; the low- density lipoprotein (LDL) receptor (LDL- R) [26]; or the asialoglycoprotein receptor (ASGP- R) [27]. It is likely that several molecules form a receptor complex at the cell surface that interacts with the HCV particle. After attachment and receptor- mediated endocytosis, the HCV nucleocapsid is released in the cell cytoplasm as a result of viral and cellular membrane fusion mediated by HCV surface glycoproteins [9,28]. Each of the early steps of HCV infection represents a potential target for anti- HCV drugs, including receptor attachment, receptor- mediated endocytosis, and fusion. In the absence of appropriate study models, no such drugs have been developed so far. However, retroviral pseudoparticles harboring folded HCV envelope glycoproteins [9,10] and the first generation of HCV- productive cell culture systems [16- 18] should facilitate their development.
3.2. Polyprotein synthesis
Decapsidation of viral nucleocapsids liberates free genomic RNAs in the cell cytoplasm, where they serve, together with newly synthesized RNAs, as messenger RNAs for synthesis of the HCV polyprotein. HCV genome translation is under the control of the internal ribosome entry site (IRES), located within the 5' untranslated region of the genome and the first nucleotides of the core- coding region [29]. The HCV IRES has the capacity to form a stable pre- initiation complex by directly binding the 40S ribosomal subunit [30,31]. The 40S subunit assembles with eukaryotic initiation factors and the initiator tRNA to form a 48S particle which then associates with the 60S ribosomal subunit to form a functional 80S ribosome that initiates viral protein synthesis [32]. There is evidence that a number of cellular proteins interact with the HCV IRES, including the polypyrimidine tract- binding protein [33]. In addition, a frameshift protein, called F protein, is synthesized from the core- coding region at an unknown frequency and yield.
Various antiviral approaches have been developed to target the HCV IRES and polyprotein translation. Nucleic acid- based strategies, including ribozymes, antisense oligonucleotides, RNA aptamers or silencing RNAs have been shown to potently inhibit HCV IRES in various in vitro systems [34,35]. These approaches have failed to significantly inhibit HCV replication in vivo so far [36]. One explanation could be the fact that nucleic acid- based therapies target the IRES RNA structure, which is not present as such within infected cells. The HCV IRES has a complex three- dimensional structure involving the IRES RNA, ribosomal subunits, and both canonical and non- canonical cellular factors. New drug design approaches based on three- dimensional mapping of the target functional structures in the HCV IRES are currently being developed.
3.3. Post- translational processing
HCV genome translation generates a large precursor polyprotein, which is secondarily co- and post- translationally cleaved by means of cellular and viral proteases into at least 10 viral proteins. Cleavage by host peptidases yields the core protein, E1 and E2 envelope glycoproteins, and the small p7 protein [29]. It has been suggested that p7 could form ion channels in the endoplasmic reticulum membrane, and that this may be necessary to HCV replication. It therefore provides a potential target for antiviral intervention.
The zinc- dependent NS2- 3 auto- protease ensures cis- cleavage of non- structural proteins NS2 and NS3. No NS2 protease inhibitors have been developed so far.
NS3 needs to assemble with its cofactor NS4A to catalyze cis- cleavage at the NS3- NS4A junction and trans- cleavage at all downstream junctions including NS4A- NS4B, NS4B- NS5A, and NS5A- NS5B [29]. The NS3 protease represents an excellent target for new anti- HCV drugs. Extended contacts are made between the polyprotein and NS3 protease during recognition of polyprotein processing sites. Contact regions provide potential inhibitor binding sites on NS3. These include the protease active site, which is targeted by a large number of molecules currently at the preclinical or early clinical developmental stage; the polyprotein substrate recognition sites, also targeted by molecules in preclinical development; the zinc- binding site; the region of interaction with the NS4A cofactor [37]. The use of NS3 proteinase inhibitors could reinforce the effect of interferon alfa when both drugs are administered simultaneously. Indeed, recent evidence suggests the NS3/NS4A complex catalyzes the cleavage of the Toll- like receptor 3 adaptor protein TRIF, leading to the inhibition of the interferon regulatory element 3 (IRF- 3) [38,39]. NS3 proteinase inhibition could thus (pending experimental demonstration) restore interferon responses in cells not responding to interferon alfa.
3.4. HCV replication
HCV infection leads to the formation of a replication complex that associates viral proteins, cellular components, and nascent RNA strands. The membranous web is derived from endoplasmic reticulum membranes and consists of small vesicles embedded in a membranous matrix, forming a membrane- associated multiprotein complex that contains all of the non- structural HCV proteins [40]. HCV replication is catalyzed by the NS5B RNA- dependent RNA polymerase (RdRp). The positive- strand genome RNA acts as a template for the synthesis of a negative- strand intermediate of replication. Negative- strand RNA then serves as a template to produce numerous strands of positive polarity that will subsequently be used for polyprotein translation, synthesis of new intermediates of replication, or packaging into new virus particles [29]. Viral and cellular factors are required for HCV replication: indeed, the NS3 NTPase/helicase and the NS5A protein play a role in viral replication, as does the polypyrimidine tract- binding protein.
The HCV RdRp displays the classical Finger/Palm/Thumb motif observed in nucleic acid polymerases and a number of interaction sites that can be targeted by specific inhibitors [37]. It is, however, unique with its fully encircled active site into which nucleotides can bind in the absence of the template. The active site of the enzyme is a target for nucleoside/nucleotide analogue inhibitors. In addition, various sites have been identified for non- nucleosidic inhibitors, which induce conformational changes by their fixation that alter RdRp structure and function. A large number of HCV RdRp inhibitors are currently at the preclinical developmental stage.
The NS3 helicase's primary function is to unwind the viral genomic RNA during replication. It is also a potential target for specific inhibitors. However, no such molecule has yet reached the clinical developmental stage. Although its actual role and its three- dimensional structure remain largely unknown, NS5A also represents a potential target for antiviral intervention.
3.5. Virus assembly and release
Because of the lack of appropriate study models, little is known about virus assembly and release. Viral particle formation is probably initiated by the interaction of the core protein with genomic RNA, and the core- RNA interaction could play a major role in the switch from replication to packaging. Virus assembly could occur in the endoplasmic reticulum, and both the endoplasmic reticulum and the Golgi apparatus could be involved in the late maturation steps [41]. The mechanisms underlying exportation of mature virions in the pericellular space remain unknown. No inhibitors of the late steps of the HCV lifecycle have been developed so far, but the recent development of a productive cell culture system and the foreseeable understanding of these mechanisms could accelerate the development of such molecules.
4. Recent clinical hepatitis C drug developments
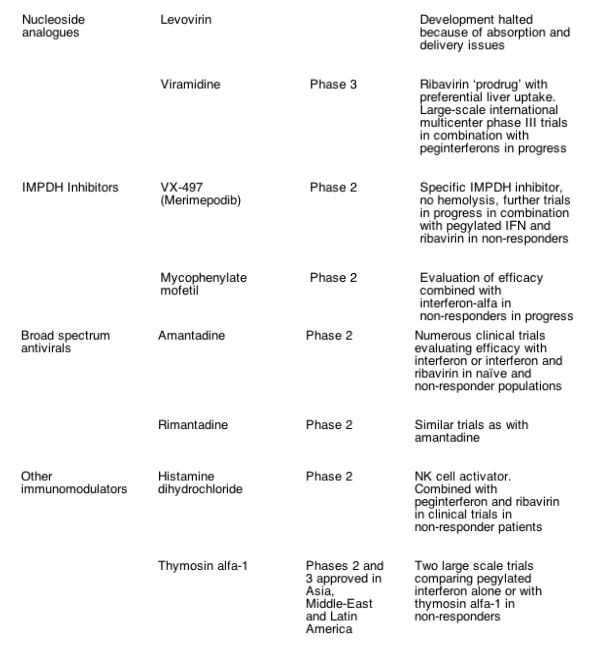
Advances in in vitro HCV infection systems and knowledge of HCV virology have resulted in the development of a number of novel compounds that are now being evaluated in early- and later- phase clinical trials for their efficacy in treating HCV (Table 1). Most current compounds in clinical trials were developed with the goal of either disrupting viral replication machinery, or modulating the antiviral host immune response. The following compounds in clinical development may prove to be the next viable alternatives or adjuncts to current therapeutic regimens.



4.1. HCV specific protease inhibitors
The NS3- 4A region comprises a multifunctional protein that contains a serine protease at the N- terminus and a cofactor NS4A that cleaves the viral polyprotein into functional proteins essential for replication and may also reduce the innate immune response by the host cell. The substrate- binding cleft of this region is shallow, which posed initial difficulties in developing specific enzyme inhibitors. BILN- 2061 (Boehringer- Ingelheim, Ingelheim, Germany) was the first HCV protease inhibitor to enter clinical trials, and it demonstrated a transient but significant and rapid decline in HCV RNA levels in HCV genotype- 1 infected patients that were less impressive in patients with non- genotype 1 infection [42- 44]. However, further development of this compound was halted because of cardiotoxicity issues in laboratory animals given supratherapeutic doses for several weeks. Interestingly, the observation that single mutations could confer resistance in vitro to this compound indicated that escape mutations were likely with this class of drug when used as a monotherapy.
Other NS3- 4A serine protease inhibitors have advanced to the clinic. VX- 950 (Vertex Pharmaceuticals, Inc., Cambridge, MA, USA) is a specific, peptidomimetic inhibitor of NS3- 4A protease that includes an ��- ketoamide moiety that anchors at the active site. VX- 950 has demonstrated antiviral activity in HCV replicon assays, even against BILN- 2061 resistant strains [45]. Early phase clinical data suggested the compound was well tolerated and orally bioavailable. In a phase Ib study, HCV genotype- 1 infected patients were dosed for 14 days with 450mg, 750mg VX- 950 every 8h, 1250mg every 12h, or placebo. All 28 patients that received VX- 950 had at least a 2- log10 decrease in HCV RNA within 72h. The highest trough plasma concentrations of VX- 950 were observed in the 750- mg dose group, which had a median 4.4- log10 reduction in HCV RNA levels [46]. Four patients in the higher dose group achieved undetectable HCV RNA levels during the study. The drug appeared to be well tolerated, with no additional safety or cardiac concerns at this stage. Further efficacy and pharmacokinetic data are awaited. These encouraging results have led to the development of phase II clinical trials with VX- 950 alone or in combination with pegIFN- alfa and/or ribavirin that are underway.
The protease inhibitor SCH 503034 (Schering Plough, Kenilworth, NJ) is an orally active protease inhibitor that has specific antiviral activity in HCV replicon assays. In an ongoing phase II study of HCV genotype- I infected patients who were previous non- responders to pegIFN- alfa, 400mg SCH 503034 thrice daily alone was associated with a mean HCV RNA reduction of 2log10 from baseline [47]. Further phase II clinical trials in combination with pegIFN with and without ribavirin are in progress, and the results eagerly awaited.
4.2. Polymerase inhibitors
The NS5B region encodes a unique HCV RdRp that is essential for HCV replication, thus allowing for selective inhibitor development. There are several potential and favourable different binding sites on the HCV polymerase, and the use of high- throughput screening and rational drug design has led to the identification of a number of nucleoside and non- nucleoside analogues as well as thumb site HCV RdRp inhibitors [48].
NM283 (Valopicitabine, Idenix Pharmaceuticals, Cambridge, MA) is a nucleoside analog that induced a dose- dependent 1.2- log10 decline in HCV RNA in HCV genotype- 1 infected patients receiving 800mg daily for 2 weeks [49]. This compound is currently being studied in a phase IIb trial of 190 HCV genotype- 1 infected patients that failed to achieve early viral response. Following encouraging 4- week safety and efficacy data, patients are receiving NM283 alone, NM283 and pegIFN- alfa, or pegIFN- alfa and ribavirin for 48 weeks. Interim results based on 12 weeks of treatment reveal a mean HCV RNA reduction of 2.8log10 among patients receiving NM283 and pegIFN- alfa, including undetectable levels in a significant proportion of patients [50].
Non- nucleoside inhibitors of HCV RdRp include JTK- 003 and JTK- 109 (Japan Tobacco Incorporated, Tokyo, Japan). JTK- 003 appears to have advanced to phase II clinical trials in Japan for HCV genotype- 1 infected non- responder patients. Several other HCV polymerase allosteric inhibitors are currently in pre- clinical and early clinical development, but details of these compounds are not publicly available. Although these RdRp inhibitors are specific for HCV and have acceptable safety and oral bioavailability, they appear less potent, at least in terms of viral decline, than the NS3- N4A protease inhibitors and are likewise theoretically susceptible to resistance development.
A significant step forward in our current therapeutic options will occur if these drugs used alone, or in combination, can maintain their safety and efficacy profile over a prolonged dosing period and shorten the duration of treatment without the development of clinically meaningful resistance. The protease and polymerase inhibitors do offer the potential for multiple- targeted therapy that may be required in an increasingly difficult- to- treat HCV- infected population. A significant reduction in HCV RNA levels prior to or during the initials stages of current standard of care treatment would also intuitively appear beneficial. Resistance issues related to individual drugs, cross- resistance between compounds, resistance in the presence of pegIFN and ribavirin, and the clinical significance of escape mutants will need to be carefully evaluated. How these compounds will be combined in different patient populations will also require careful thought and planning to maximize the potential benefit.
4.3. Immune modulators
4.3.1. Interferons
IFN- alfa has been used for more than a decade to treat patients with hepatitis C, and this is the only class of drugs known to result in sustained viral clearance and improvement in liver histology. Various modifications to the therapeutic proteins of IFN have improved the half- life of the drug, leading to longer dosing intervals and increased response rates [2,3]. A fusion protein of IFN alfa- 2b and human serum albumin, Albuferon (Human Genome Sciences, Rockville, MD, USA), is currently being evaluated in phase II trials for safety and efficacy at 2- to- 4 week dosing intervals [51].
Several other genetically modified or alternative IFN compounds are being evaluated (Table 1), but they are likely to have similar delivery issues and side- effect profiles that are common with this class of immunomodulators, thus precluding their use in a significant proportion of patients.
4.3.2. Toll- like receptor agonists
Modulation of the host innate and adaptive immune responses through Toll- like receptors (TLR) expressed by activated immune cells has received attention recently. CPG- 10101 (Actilon, Coley Pharmaceutical Group, Wellesley, MA) is a synthetic oligodeoxynucleotide and the first of a new class of Toll- like receptor 9 (TLR9) agonists that also induces T- Helper type 1 cytokine responses, resulting in high levels of type 1 IFN, NK cell stimulation, and other viral- specific immunomodulatory responses. In a recent phase Ib clinical trial of CPG 10101, HCV genotype- 1 infected patients who received at least 1mg CPG 10101 twice weekly for 4 weeks experienced increases in IFN- alpha and other markers of immune response along with a mean 1- log10 decline in HCV RNA levels [52]. Ongoing phase II studies are evaluating this strategy in prior relapse and non- responder patient populations in combination with standard of care therapy.
ANA245 (Isatoribine, Anadys Pharmaceuticals, San Diego, CA, USA) is a TLR- 7 agonist that resulted in an 88% median decline in HCV RNA levels after intravenous dosing of 800mg daily for 1 week [53]. Preliminary 28- day safety and efficacy data are awaited for ANA975, an oral prodrug of isatoribine. As the immune responses are generated to endogenously derived peptides these compounds are expected to have a reduced systemic side effect profile compared with current IFN therapy, and fewer resistance issues than direct viral targeted therapy.
4.3.3. Ribavirin analogues
Viramidine (Valeant Pharmaceuticals, Costa Mesa, CA) is an inactive prodrug of ribavirin that is activated through deamination in the liver by adenosine deaminase [54]. Preferential viramidine delivery to the liver and poor uptake in erythrocytes results in reduced hemolytic anemia. Results from a phase II proof- of- concept study of viramidine in combination with pegIFN- alfa in 180 treatment- naive HCV patients indicates similar antiviral efficacy at week 24 of therapy compared with the standard combination therapy (37 vs. 44%), but significantly less anemia (4 vs. 27%) for the 600- mg- daily group [55]. This drug is being evaluated in phase III clinical trials, and although it may not show an incrementally higher virologic response compared with existing combination therapy, improvements in the safety profile over ribavirin would represent important clinical progress and would likely increase the proportion of patients currently eligible for therapy.
4.4. RNA- based therapy
Antisense oligonucleotides, ribozymes, and more recently small interfering RNAs (siRNA) have all been explored as potential antiviral therapies. However, due to their size and chemical composition, these compounds are not orally bioavailable and require parenteral administration. Limited efficacy and adverse effects hampered early phase clinical trials with ISIS- 14803 (ISIS Pharmaceuticals, Inc., Carlsbad, CA) and Hepatazyme (Ribozyme Pharmaceuticals, Inc., Boulder, CO or Sirna Therapeutics, Inc., San Francisco, CA). Methods for effective in vivo drug delivery for siRNAs are being explored. Chemically modified siRNA appears to inhibit HCV in tissue culture, and the use of a multi- targeting approach has been adopted to develop siRNAs active against HCV IRES, NS3 protease, and NS5b polymerase in pre- clinical studies (Benitec, Inc., Mountain View, CA). This approach is expected to reduce emergence of viral resistance, but achieving targeted drug delivery for these compounds remains a major obstacle.
4.5. Vaccines
Cellular immune responses to the HCV E1 envelope protein are frequently suppressed or absent in patients with chronic hepatitis C. A clinical- grade HCV E1 protein produced and purified from mammalian cells has been evaluated in initial trials. In a phase IIa study, most patients developed a strong E1- specific T- helper response [56]. After the second course of E1 injections, levels of anti- E1 antibodies increased 3- to 4- fold, and the proportion of patients with a significant T cell response to E1 increased from 9 to 91%. However, these cellular immune responses were not accompanied by any significant reductions in serum HCV RNA.
IC41 (Intercell AG, Vienna, Austria) is a synthetic vaccine incorporating five peptides that include several cytotoxic and T- helper epitopes. In a phase II study, IC41 was well- tolerated and capable of inducing IFN- gamma cytotoxic and T- helper immune responses in prior non- responders to standard therapy [57].
|
|
| |
| |
|
|
|