| |
Metabolics/Belly Fat Advance HCV Disease
|
| |
| |
"Impact of obesity on treatment of chronic hepatitis C"
Hepatology June 2006, Vol 43, Issue 6, pages 1177- 1186
".....Current understanding of obesity is that it constitutes a metabolic condition and is not simply a function of having high body weight. .....clinical factors associated with the metabolic syndrome - including elevated waist- to- hip ratio, the presence of insulin resistance, and diabetes - have been shown to predict more advanced forms of chronic hepatitis C.... Several studies have demonstrated that SVR rates are lower in patients with coexistent insulin resistance and/or hepatic steatosis/steatohepatitis.... HCV RNA levels have been found to correlate with degree of steatosis, viral eradication results in regression of steatosis, and steatosis returns with relapse....TREATMENT STRATEGIES: .......the most direct approach is to encourage weight loss and exercise before treatment..... Recently reported data suggest that treatment with the insulin- sensitizing medications metformin and the thiazolidinediones (pioglitazone and rosiglitazone) reduced serum ALT and histologic features of hepatic steatosis, inflammation, and fibrosis, as well as increased insulin sensitivity, in nondiabetic patients with nonalcoholic steatohepatitis....... Other approaches to enhance response to combination treatment with peginterferon alpha plus ribavirin include longer duration of treatment and, possibly, higher flat doses, which may counteract the decreased bioavailability of drug and increased resistance to interferon- based therapies. Longer treatment, especially in patients infected with HCV genotype 1, may enhance SVR rates. In addition, higher doses of peginterferon may enhance SVR rates in obese individuals. Rather than being based strictly on weight, higher peginterferon doses could be based on a threshold BMI (e.g., 30 kg/m2) or a threshold value for visceral fat or insulin resistance, as measured HOMA- IR index....."
Authors: Michael R. Charlton 1 *, Paul J. Pockros 2, Stephen A. Harrison 3
1Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, Rochester, MN
2Division of Gastroenterology/Hepatology, Scripps Clinic, La Jolla, CA
3Division of Gastroenterology and Hepatology, Brooke Army Medical Center, Fort Sam Houston, TX
Potential conflict of interest: Dr. Pockros is on the speakers' bureau for and received grants from Roche, Valeant, Bristol- Meyers- Squibb, and Gilead. He advises Roche and Valeant. Dr. Charlton is a consultant for Bristol- Meyers- Squibb and has received research funding from Roche.
Funded by:
Roche Laboratories, Inc
Abstract
Obesity and the metabolic syndrome have hepatic manifestations, including steatosis (fatty liver) and progression of fibrosis. In individuals with chronic hepatitis C, obesity is associated with inflammation, insulin resistance, steatosis, progression of fibrosis, and nonresponse to treatment with interferon or peginterferon alpha and ribavirin. Patients with both hepatitis C and obesity- related nonalcoholic fatty liver disease are at greater risk for more advanced liver disease. We review the mechanisms by which obesity may be associated with decreased efficacy of interferon- based therapies in individuals with chronic hepatitis C and the therapeutic strategies that may increase the effectiveness of these therapies in obese individuals.
I. Introduction
Recent data from the National Health and Nutrition Examination Survey (NHANES) indicate that approximately 30% of adults in the United States are obese, as defined by a body mass index (BMI) 30 kg/m2.[1][2] It has been estimated that approximately 20% of individuals infected with the hepatitis C virus (HCV) are obese and that obesity in these individuals is associated with steatosis and the progression of fibrosis.[3- 7] These factors are associated with nonresponse to treatment with interferon or peginterferon alpha and ribavirin.[3][6] We review the mechanisms by which obesity may be associated with decreased efficacy of interferon- based therapies in individuals with chronic hepatitis C (CHC) and the therapeutic strategies that may increase the effectiveness of these therapies in obese individuals.
II. Obesity, the Metabolic Syndrome, and Hepatitis C
A. The Metabolic Syndrome
The metabolic syndrome is a compilation of clinical factors thought to have a common denominator in insulin resistance. Some disagreement exists as to the specific characteristics that define the metabolic syndrome, but the National Cholesterol Education Program - Adult Treatment Panel III (NCEP- ATP III) guidelines are widely used and include three or more of the following[2]:
- Waist circumference >102 cm (40 in) for men or >88 cm (35 in) for women (abdominal [i.e., visceral] obesity)
- Serum high- density lipoprotein (HDL) cholesterol <40 mg/dL for men or <50 mg/dL for women
- Serum triglycerides >/= 150 mg/dL
- Systolic blood pressure >/= 130 mm Hg or diastolic blood pressure 85 mm Hg
- Fasting glucose >/= 110 mg/dL
Another set of criteria has been proposed by the World Health Organization (WHO)[8] and includes diabetes, increased insulin levels, increased fasting serum glucose, or elevated postmeal glucose alone with at least two of the following:
- Waist- to- hip ratio >0.9 for men or >0.85 for women, BMI >30 kg/m2, or waist circumference >37 cm
- Triglycerides >/= 150 mg/dL or HDL <35 mg/dL for men or <39 mg/dL for women
- Blood pressure >/= 140/90 mm Hg
- Microalbuminuria >/= 20 g/min or albumin/creatinine ratio >/= 30 mg/g
More recently, the International Diabetes Federation (IDF) proposed a revised definition that focused on central obesity based on ethnicity and an additional two of the following four parameters[9]:
- Triglycerides >/= 150 mg/dL or on medication
- HDL <40 mg/dL for men or <50 mg/dL for women or on medication
- Blood pressure >/= 130/85 mm Hg or on medication
- Fasting glucose >/= 100 mg/dL or previously diagnosed with diabetes
The significance of central obesity is evident in all three sets of criteria, because approximately 60% of patients who have the metabolic syndrome will be obese.[10] However, these three definitions differ with regard to (1) the appropriate cutoff for blood pressure; (2) the inclusion of diabetics in the National Cholesterol Education Program (NCEP) and WHO, but not the IDF criteria; and (3) the inclusion of microalbuminuria in the WHO criteria. There are questions regarding all three sets of criteria, because it is unclear if all parameters are weighted equally or if some are more important than others. Furthermore, there are questions regarding whether other risk factors (e.g., C- reactive protein or adiponectin levels or family history of diabetes or heart disease) should be included.[11]
T metabolic syndrome has been estimated to affect approximately 34% of U.S. adults according to the NCEP criteria and 39% according to IDF criteria, and its incidence has been increasing in parallel with the increasing incidence of obesity.[12][13] Although the prevalence of the metabolic syndrome among hepatitis C- infected patients worldwide is unknown, the prevalence of overweight or obesity among patients with hepatitis C has been estimated to be between 17% and 38% in China, Western Europe, and North America.[7][14- 16]
Although earlier studies in hepatitis C- infected patients used body weight as a measure of obesity, this was found not to be as accurate as BMI, which correlates better with total body fat content.[3][17] BMI, however, also has limitations as a measure of obesity, because fat distribution and the possibility of fluid retention are not considered when determining BMI.[18] Therefore, newer guidelines, such as those for the metabolic syndrome, are focusing more on central adiposity as determined by waist- to- hip ratio.
B. Hepatic Manifestations of the Metabolic Syndrome
Among the specific hepatic manifestations of the metabolic syndrome that may exacerbate HCV infection is nonalcoholic fatty liver disease (NAFLD). This disease has a clinical spectrum of histologic changes ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), which may progress further to cirrhosis and/or hepatocellular carcinoma.[5][19- 21] It has been estimated that NAFLD affects approximately 45% of Hispanic adults and 33% of Caucasian adults in the United States and that there is a strong association between NAFLD and obesity.[22][23]
Individuals with coexistent CHC and NAFLD, irrespective of the presence of NASH, are much more likely to develop more advanced liver disease than those without HCV infection.[5] Coexistent hepatic steatosis is common in CHC, occurring in more than 50% of patients, but it is usually mild.[5][19][24] Obese CHC patients, however, are at greater risk for the development of coexistent steatosis and more advanced liver disease.[5][19][24] Whereas both viral and host factors contribute to coexistent steatosis, clinical factors associated with the metabolic syndrome - including elevated waist- to- hip ratio, the presence of insulin resistance, and diabetes - have been shown to predict more advanced forms of CHC.[25][26]
C. Hepatitis C and Pegylated Interferons
The current standard of care for the treatment of chronic HCV infection is combination therapy with peginterferon alpha and ribavirin.[27] Although its mechanisms of action are not well understood, interferon alpha may contribute to viral eradication by inducing virally infected hepatocytes to manufacture proteins that interfere with viral replication, as well as having more indirect immunomodulatory and anti- inflammatory effects.[28] The effects of immunomodulatory drugs such as interferon alpha are dependent on host nutritional and metabolic conditions, as well as on the ability of the host immune system to respond.[29] Attachment of interferon alpha to a large, chemically inactive polyethylene glycol (PEG) molecule has been found to reduce the rate of elimination of interferon alpha by the kidneys and to decrease its proteolytic degradation and antigenicity, thereby increasing the amount of time that the interferon is effective.[30] Currently, there are two commercially available forms of pegylated interferons: peginterferon alpha- 2b (12 kd), a covalent conjugate of a linear 12- kd PEG moiety to recombinant interferon alpha- 2b; and peginterferon alpha- 2a (40 kd), a covalent conjugate of a branched 40- kd PEG molecule to recombinant interferon alpha- 2a.[31][32]
Ribavirin, a nucleoside analog exhibiting antiviral activity against a variety of RNA viruses, is used in combination with pegylated interferon to treat hepatitis C.[33][34] The mechanisms of action of ribavirin are not fully known but may include inhibition of inosine monophosphate dehydrogenase, immunomodulatory effects, and, more likely, its mutagenic activity toward RNA viruses.[35][36] Current evidence suggests that a flat dosage of ribavirin at 800 mg/d in combination with peginterferon for 24 weeks is effective in treating patients infected with HCV genotypes 2 and 3, whereas weight- based doses of ribavirin (1,000 mg/d for patients weighing 75 kg and 1,200 mg/d for patients weighing >75 kg) plus peginterferon for 48 weeks result in better SVR rates for patients infected with HCV genotype 1, particularly in patients who have a high viral load.[37][38] Newer evidence suggests that even higher doses of ribavirin may result in better overall response rates in patients infected with HCV genotype 1, but this remains controversial.[38][39]
Treatment with peginterferon plus ribavirin has been found to induce a sustained virologic response (SVR), defined as undetectable HCV RNA 6 months after the end of treatment, in approximately 50% of patients.[33][34][37] SVR rates are independently associated with several viral factors, including viral genotype and pretreatment viral load, although the most powerful predictor of SVR is viral genotype.[33][37] In addition, host factors such as increased BMI, insulin resistance, African American race, and hepatic steatosis/steatohepatitis or fibrosis are also associated with lower SVR rates, although to a lesser extent than viral genotype and viral load.[3][24][40- 43] Although BMI is inversely correlated with SVR, body weight by itself has been shown to be, at best, a relatively weak predictor of response to peginterferon plus ribavirin.[3][33][42][44]
III. Obesity and SVR
Several mechanisms have been proposed relating obesity to decreased rates of SVR in response to treatment with peginterferon plus ribavirin in individuals with hepatitis C (Fig. 1).[18] The first mechanism hypothesizes that obesity is an inflammatory condition, resulting in an abnormal immune response to therapy. The second mechanism hypothesizes that obesity causes insulin resistance and hepatic steatosis, which can lead to steatohepatitis and hepatic fibrosis, resulting in direct or indirect interference with the effect of interferon on hepatocytes. A third mechanism hypothesizes that obesity results in decreased bioavailability of peginterferon alpha. We review the evidence for each of these proposed mechanisms, with the aim of gaining a better understanding of how obese CHC patients can be treated more effectively.
A. Obesity as an Inflammatory Condition That Can Indirectly Interfere With the Immunomodulatory Effects of Peginterferon alpha
White adipose tissue is metabolically active and is not just a storage depot for fat.[45] Adipose tissue secretes many proteins, including adipokines, which regulate hepatic and peripheral glucose and lipid metabolism.[45][46] These proteins also modulate immune responses that can act as pro- and anti- inflammatory factors, although some of the specific roles of these proteins are still being studied.[47][48]
One of the adipokines secreted by adipose cells is leptin. Increased levels of leptin are associated with obesity and steatosis, although leptin resistance or relative hypoleptinemia may also occur.[49] Leptin has proinflammatory properties in vitro, primarily by upregulating the production of several important proinflammatory T h1 cytokines - including interleukin (IL)- 1beta, IL- 6, IL- 12, and tumor necrosis factor a (TNF- a) - and decreasing the production of the anti- inflammatory cytokine IL- 10.[50][51] In dendritic cells, one of the most important types of antigen- presenting cells, leptin increases the production of IL- 1, IL- 6, IL- 12, TNF- , and macrophage inflammatory protein (MIP)1a and decreases the production of IL- 10, thus stimulating heterologous T h1 responses.[52] Furthermore, leptin has antiapoptotic effects on dendritic cells.[52] Thus, leptin stimulates heterologous T h1 responses and inhibits heterologous T h2 immune responses.[53] Because T h 1 responsiveness has been shown to be critical to SVR in HCV- infected patients treated with peginterferon, alone or in combination with ribavirin, it is likely that leptin resistance rather than hyperleptinemia contributes to decreased SVR in obese patients.[54]
Leptin also appears to play a role in the activation of multiple signaling pathways in hepatic stellate cells, further contributing to intrahepatic inflammation and fibrogenesis. Recent evidence suggests that leptin enhances the activation of nuclear factor kB and the expression of monocyte chemoattractant 1 (MCP- 1) and vascular endothelial growth factor (VEGF). In vitro exposure of hepatic stellate cells to leptin resulted in upregulated expression of MCP- 1 and VEGFmRNA and protein.[55] Moreover, in vivo administration of leptin to Ob mice during chloroform- induced acute liver injury increased MCP- 1 expression, inflammation, and necrosis, all of which were less severe in leptin- deficient mice.[55] Taken together, these findings suggest another possible mechanism by which nonresponse to therapy is enhanced in obese patients.
There are also associations between resistin, IL- 6, and adiponectin levels and obesity.[56] Whereas resistin and IL- 6 were found to be increased in the setting of obesity, the plasma concentrations of the proinflammatory protein leptin and the anti- inflammatory protein adiponectin were shown to vary inversely in nondiabetic normal- weight and obese women.[57] Plasma adiponectin concentration showed a strong positive association with insulin sensitivity and a strong negative association with TNF- a secretion, and it may inhibit Kupffer cell activation and further release of TNF- a.[58][59] Decreased plasma concentrations of adiponectin are associated with obesity, dyslipidemia, insulin resistance, and NAFLD.[57][60][61] The role of adiponectin in patients with hepatitis C is not fully defined, however. Whereas some studies have suggested that adiponectin is negatively correlated with BMI and steatosis, others have not demonstrated this correlation, or have only noted the association with steatosis in male patients.[62- 64] Data in patients with NAFLD indicate that hypoadiponectemia is associated with more extensive inflammation.[48] However, two studies have suggested that adiponectin is positively correlated with necroinflammation in patients with hepatitis C - although these studies comprised mostly normal- weight patients infected with HCV genotype 3 who demonstrated less insulin resistance than patients infected with HCV genotype 1.[63][65] Another proinflammatory cytokine, TNF- , is upregulated in obese patients with NAFLD and in patients with hepatitis C.[48][66][67] In addition to interfering with insulin signaling, increased levels of TNF- a are associated with a decreased response to antiviral therapy.[66]
Ultimately, increased visceral obesity is associated with an imbalance in adipokine production, with increased production of the proinflammatory cytokines IL- 6 and TNF- a, diminished production of adiponectin, and increased insulin and leptin resistance.[68] This results in chronic inflammation, subsequent increased oxidative stress, and decreased response to Iinterferon alpha, which may be due, in part, to inhibition of the JAK/STAT pathway.[69] Further studies are needed to clarify the exact mechanisms through which this altered cytokine milieu affects innate immunity in humans.
B. Insulin Resistance Is Associated With Steatosis, Fibrosis, and Impaired Response to Combination Therapy.
Insulin resistance is commonly seen in patients with hepatitis C. In fact, epidemiological studies suggest the prevalence of diabetes mellitus is several- fold higher among patients with hepatitis C than controls and vice versa.[70] Insulin resistance may be mediated by both viral and host interactions. Consequences of insulin resistance in patients with hepatitis C include the development of hepatic steatosis and liver fibrosis, as well as higher baseline viremia.[71]
The development of insulin resistance in HCV- infected patients is thought to be due to a combination of both host- and virus- mediated pathways. Host- related factors typically seen in patients with NAFLD - including overweight/obesity, decreased physical activity, older age, and diets high in saturated and trans- fatty acids or fructose - are thought to contribute to insulin resistance.[72][73] Experimental data suggest that certain protein components of HCV - specifically the core and NS5A proteins - may also induce insulin resistance directly and that this occurs early in the course of infection.[74] TNF- a may mediate this process and has been shown to be upregulated in patients with hepatitis C.[66] In addition, insulin- resistant, transgenic mice infected with hepatic C core protein and treated with anti- TNF- a have shown improved insulin sensitivity.[74] Interestingly, HCV has been shown to upregulate expression of the protein suppressor of cytokine signaling 3 (SOCS- 3), a negative regulator of insulin signaling, which results in inhibition of phosphorylation of insulin receptors and enhancement of insulin resistance.[75][76] In patients infected with HCV genotype 1, nonresponse to treatment has been associated with obesity and increased hepatic expression of SOCS- 3.[77]
Hepatic steatosis in patients with CHC is a common finding. Approximately 55% of these patients develop some degree of hepatic steatosis, but the majority (75%) will have less than 30% parenchymal involvement.[78] Insulin resistance may be the cause, rather than the consequence, of hepatic steatosis in individuals infected with HCV, particularly those with genotype 1 infection (Fig. 2).[79][80] Under conditions of insulin resistance and obesity, hepatic regulatory mechanisms may be unable to prevent fat accumulation in hepatocytes, thus exacerbating chronic HCV infection and disease progression.[81] No fewer than 11 reports in the literature have linked overweight or obesity to hepatic steatosis in patients with CHC.[78] A recent study from Spain found that 34% of patients with coexistent NASH and CHC were obese, whereas no obesity was observed in patients without evidence of NASH.[82]
Nonadipocytes, including hepatocytes, have a limited capacity to store excess fat. When exposed to high concentrations of plasma lipids, such as long- chain fatty acids, these cells may develop steatosis, become functionally deficient (lipotoxicity), and ultimately undergo programmed cell death (lipoapoptosis).[83- 86] Leptin and possibly adiponectin produced by adipocytes may have antisteatotic and antiapoptotic effects on nonadipocytes.[87- 89] Although visceral obesity is characterized by increased serum leptin concentrations, this is frequently accompanied by relative hypoleptinemia and/or leptin resistance.[86] Thus, in individuals who are both obese and infected with HCV, enhanced serum leptin concentrations may lead to the progression of steatosis to steatohepatitis, and more severe liver damage may be associated with the combination of lipotoxicity and the additional oxidative stress associated with viral infection (the second hit hypothesis).[90][91] This is supported by findings that grade of steatosis was significantly associated with BMI and was predictive of more severe levels of fibrosis in HCV- infected patients.[92]
Evidence suggests that genotype 3 infection may lead to the development of hepatic steatosis regardless of insulin resistance.[93] HCV RNA levels have been found to correlate with degree of steatosis, viral eradication results in regression of steatosis, and steatosis returns with relapse.[80] A review on the mechanisms behind virally mediated steatosis has recently been published.[78]
Hepatic fibrosis may occur as a result of obesity, host- and virus- mediated insulin resistance, and hepatic steatosis, and there is a direct link between obesity and fibrosis.[7] Among patients with normal serum alanine aminotransferase levels, only BMI has been shown to be a predictor of advanced disease.[7] Insulin resistance has been shown to be associated with increased fibrosis, and the degree of insulin resistance correlates with progression of fibrosis.[94] Numerous studies have further demonstrated an association between hepatic steatosis and fibrosis, but recently this has been questioned.[95] The mechanism linking fatty infiltration and insulin resistance with collagen deposition is thought to involve the stimulation of hepatic stellate cells. For example, high glucose concentrations and hyperinsulinemia have been shown to increase the expression of connective tissue growth factor (CTGF) in hepatic stellate cells, and the expression of CTGF is elevated in human liver biopsy specimens from patients with nonalcoholic steatohepatitis and in a rat model of obesity and type 2 diabetes.[96] In nondiabetic CHC patients, insulin resistance is associated with level of fibrosis.[94] Furthermore, recent findings suggest that the mechanisms linking fatty infiltration and insulin resistance with collagen deposition involve the stimulation of hepatic stellate cells by apoptotic products and/or leptin.[97][98]
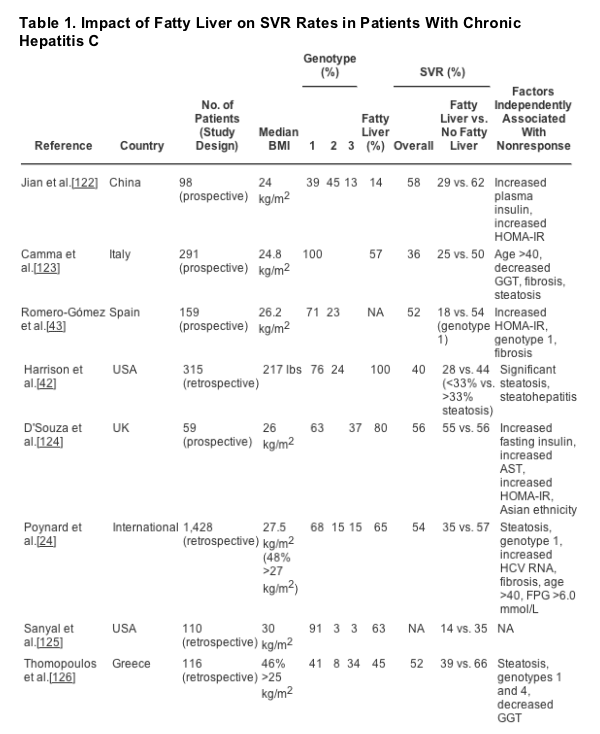
Several studies have demonstrated that SVR rates are lower in patients with coexistent insulin resistance and/or hepatic steatosis/steatohepatitis (Table 1). The pathogenic mechanisms for this are not fully understood. However, it has been demonstrated that insulin infused into HCV replicon cells results in increased amounts of HCV RNA, and this may, in part, explain the decreased effectiveness of antiviral therapy.[99] How and why this occurs is unknown, but altered effects on innate cellular immunity have been debated. Similar enzymes (kinases) are involved in activation of immune regulatory pathways and in insulin signaling. As previously noted, oxidative stress may inhibit activation of the JAK/STAT pathway,[69] which is also the signaling pathway through which downstream production of interferon- sensitive genes occurs. Further investigation is needed to decipher the specific mechanisms related to decreased antiviral effectiveness.
In HCV- infected individuals, interferon alpha is thought to induce a cascade of gene expression that leads to the synthesis of certain proteins in hepatocytes, including the enzyme 2',5'- oligoadenylate synthetase (2- 5 OAS), which can interfere with viral replication. Treatment of HCV- infected individuals with interferon, with or without ribavirin, has been shown to induce expression of this enzyme in peripheral blood mononuclear cells (PBMCs).[100- 102] After treatment with interferon, PBMCs from viremic HCV- infected patients expressed lower levels of 2- 5 OAS than those from nonviremic individuals, both before and after incubation with interferon.[103] The 2- 5 OAS response ratio, defined as the maximal 2- 5 OAS level after treatment divided by the pretreatment 2- 5 OAS level, has been found to be significantly higher in nonobese than in obese HCV- infected individuals, suggesting that the former exhibit stronger biologic responses upon exposure to exogenous interferon alpha.[104] The HCV core protein was found to reduce transcription of the interferon- activated antiviral genes 2- 5 OAS, MxA, and protein kinase R.[105] In addition, 2- 5 OAS has been shown to interact with the HCV protein NS5A and to inhibit the antiviral activity of interferon.[106]
C. Decreased Bioavailability of Interferon alpha in Obese Patients
According to this hypothesis, subcutaneous injection of peginterferon alpha into obese patients, who have higher levels of subcutaneous fat, may result in impaired drug absorption.[3] When injected subcutaneously, proteins larger than 15 kd are primarily taken up by the lymphatic system, as opposed to direct uptake by the blood circulation.[107] Lymph passes through the lymph nodes, which are the primary sites of recruitment, activation, and proliferation of immune system cells, including lymphocytes, monocytes/macrophages, and dendritic cells.[108] Because lymphatic drainage is altered in obese individuals, increased levels of fat may interfere with lymphatic uptake of peginterferon alpha; and with the amount in circulation.[109][110]
Obesity also has different effects on the two forms of peginterferon alpha. When administered to any individual, a drug has a certain volume of distribution, which has been defined as the ratio of the amount of drug in the body to the concentration of the drug in the blood or plasma.[111] The volume of distribution of peginterferon alpha- 2a is 8- 12 L/kg and is not substantially affected by body weight.[30][112] In contrast, the volume of distribution of peginterferon alpha- 2b (about 1 L/kg) has been reported to vary according to weight.[31][112] This is a consequence of the relative molecular weights of the two protein derivatives, in that peginterferon alpha- 2a has a molecular weight of approximately 60 kd (20 kd for the interferon moiety plus 40 kd for the polyethylene glycol moiety), whereas peginterferon alpha- 2b has a molecular weight of approximately 32 kd (20 kd for the interferon moiety plus 12 kd for the PEG moiety).[31][32]
Peginterferon alpha- 2b is administered on the basis of body weight (1.5 g/kg/wk), whereas peginterferon alpha- 2a is administered as a fixed dosage of 180 ug/wk. Often, drugs dosed by weight have a wide volume of distribution or a narrow therapeutic index (e.g., similar maximum tolerated and minimum effective doses).[112] The rationale for weight- based dosing is to achieve enough drug exposure to produce the intended therapeutic effect and to reduce interindividual variability.[113] Peginterferon alpha- 2a is administered in fixed dosages of 180 ug/wk, but higher fixed doses or longer treatment times may be required when disease or host conditions are associated with a lower likelihood of attaining SVR.[112] For example, this may be necessary for obese individuals, who have a lower likelihood of attaining SVR. Longer treatment times with peginterferon plus ribavirin may also be necessary.[112]
IV. Possible Treatment Strategies to Enhance Efficacy of Combination Therapy in Obese HCV- Infected Patients
In formulating more effective treatment regimens for obese patients with CHC, it is important to use current knowledge of the metabolic effects of obesity. Thus, the most direct approach is to encourage weight loss and exercise before treatment. Obesity is associated with a reduced response to combination therapy as well as increased steatosis and fibrosis.[3][114] Weight loss in HCV- infected patients, however, has been associated with a reduction in steatosis and significant decreases in fibrosis score (P = .04) and activated stellate cells (P = .004).[115] In addition, weight loss can lead to a reduction in other factors of the metabolic syndrome, including blood pressure and serum triglyceride concentration. Weight loss can also help control comorbid conditions such as mean fasting insulin concentration.[115][116]
Treatment of insulin resistance before or in combination with antiviral therapy may also lead to improvements in CHC patients. Recently reported data suggest that treatment with the insulin- sensitizing medications metformin and the thiazolidinediones (pioglitazone and rosiglitazone) reduced serum alanine aminotransferase and histologic features of hepatic steatosis, inflammation, and fibrosis, as well as increased insulin sensitivity, in nondiabetic patients with nonalcoholic steatohepatitis; however, recent human data with metformin do not appear to be as promising.[117- 120] Additional trials in larger numbers of patients - as well as in patients with hepatitis C - are necessary to determine whether treatment of insulin resistance before or during treatment with peginterferon plus ribavirin would be effective in HCV- infected patients. Such trials should evaluate multiple end points, including safety data, and efficacy end points, including improvements in insulin sensitivity, cytokine profiles, histopathology, and ultimately SVR. Ideally, a correlation between improved insulin sensitivity and innate immunity signaling pathways should be sought.
Other approaches to enhance response to combination treatment with peginterferon alpha plus ribavirin include longer duration of treatment and, possibly, higher flat doses, which may counteract the decreased bioavailability of drug and increased resistance to interferon- based therapies. Longer treatment, especially in patients infected with HCV genotype 1, may enhance SVR rates.[121] In addition, higher doses of peginterferon may enhance SVR rates in obese individuals. Rather than being based strictly on weight, higher peginterferon doses could be based on a threshold BMI (e.g., 30 kg/m2) or a threshold value for visceral fat or insulin resistance, as measured HOMA- IR index.
V. Conclusions
Current understanding of obesity is that it constitutes a metabolic condition and is not simply a function of having high body weight.
Patients who have CHC and are obese are more likely to be insulin- resistant and to have more advanced hepatic steatosis/steatohepatitis and fibrosis. These latter conditions are independent predictors of nonresponse to combination therapy with peginterferon alpha and ribavirin, and obese patients are therefore more likely to be nonresponders to combination therapy. Treatment strategies that focus on improving underlying metabolic factors associated with poor response to combination therapy are thus more likely to overcome the low SVR rates often observed in obese patients infected with HCV.
|
|
| |
| |
|
|
|