| |
Fuzeon- 48 weeks Efficacy in TORO 1 & 2: subgroup analyses
|
| |
| |
Durable Efficacy of Enfuvirtide Over 48 Weeks in Heavily Treatment-Experienced HIV-1-Infected Patients in the T-20 Versus Optimized Background Regimen Only 1 and 2 Clinical Trials
JAIDS Journal of Acquired Immune Deficiency Syndromes: Volume 40(4) 1 December 2005 pp 404-412
Nelson, Mark MA, MBBS, MRCP*; Salgo, Miklos P MD, PhD****
From the *Chelsea and Westminster Hospital, London, United Kingdom; Roche, Basel, Switzerland; ****Roche, Nutley, NJ;
Although most were extensively pretreated, the patients enrolled in the 2 TORO studies presented with a range of treatment histories and demographic and baseline characteristics, reflecting the heterogeneity of the HIV-infected patient population. Therefore, we have carried out exploratory analyses to compare treatment effects between the enfuvirtide and control groups by different factors after 48 weeks.
AUTHOR DISCUSSION
Week 24 efficacy data from the phase 3 studies TORO 1 and TORO 2 have already demonstrated the antiviral and immunologic benefit of enfuvirtide in the treatment of HIV infection.10,12 The week 48 results presented here clearly confirm the week 24 data, demonstrating a consistent and durable treatment benefit for patients in the enfuvirtide group compared with patients in the control group in terms of decrease in plasma HIV-1 RNA levels and increase in CD4 counts. In addition to the patients who maintained their week 24 response, a number of patients reached <50 HIV RNA copies/mL or <400 HIV RNA copies/mL between weeks 24 and 48. This finding underscores the need for a minimum of 48 weeks of follow-up in salvage studies. It is of note that 79% of patients in the enfuvirtide arm and 86% of patients in the control arm had a prior AIDS-defining event, highlighting the fact that the TORO population was characterized by advanced HIV disease. Given this advanced stage of HIV infection as well as the degree of previous therapeutic exposure, and even allowing for the use of a new drug class, the degree and sustained nature of the responses observed in the enfuvirtide arm are encouraging; the high proportion of patients completing 48 weeks of treatment reflects the high motivation of these patients.
The week 48 virologic and immunologic responses to enfuvirtide were evaluated by subgroup analyses for patient demographic and baseline characteristics. A benefit was consistently shown in all subgroups treated with enfuvirtide compared with OB treatment alone, in terms of reducing HIV-1 RNA levels to <400 copies/mL and increasing CD4 counts. In most cases, this benefit was statistically significant. These analyses were intended to be exploratory, because patients were not stratified by category within each subgroup and the sample sizes within some categories may have been small. Therefore, these subgroup analyses should be interpreted with caution. Nevertheless, the consistency of the increased response in the enfuvirtide group compared with the control group across all the subgroups studied lends strong support to the enfuvirtide efficacy profile.
Some general trends were observed in both treatment groups. The subgroups with a lower baseline viral load, a higher baseline CD4 count, less previous antiretroviral experience, and more active drugs in the background regimen had the greatest magnitude of benefit from treatment, irrespective of treatment group. These trends emphasize the potential greater benefit of including enfuvirtide in a new treatment regimen before existing therapeutic options are exhausted and of careful optimization of the background antiretroviral using resistance testing. Other studies have also shown a significant association between baseline GSS or PSS and the response to treatment with combinations of antiretrovirals,17,18 and the use of resistance testing to optimize the choice of antiretrovirals is currently recommended in several of the published expert HIV treatment guidelines in Europe and the United States.4,19-21 Conversely, it should be noted that reductions in antiretroviral susceptibility are rarely absolute and that genotypic algorithms and phenotypic cutoffs used to categorize agents as resistant or susceptible are not always based on clinical data. Thus, a virus with a GSS or PSS of 0 may retain a degree of susceptibility to some agents sufficient to construct an OB regimen with adequate activity to supplement enfuvirtide. In particular, for patients who can no longer achieve full viral suppression, partial viral suppression with an accompanying immunologic response and prevention of disease progression could be a realistic goal of therapy.
Irrespective of treatment arm, patients with prior experience of lopinavir/ritonavir were less likely to have a virologic response than those patients naive to lopinavir/ritonavir, reflecting the fact that their benefit from lopinavir/ritonavir had already affected the baseline viral load and CD4 count. Of those patients naive to lopinavir/ritonavir, however, those with lopinavir/ritonavir in the OB fared better than those without. At the time of study initiation, lopinavir/ritonavir was an investigational agent; thus, the patients who had previously received it would have done so in a clinical trial and would likely have been extensively treatment experienced. Indeed, further analyses have shown prior lopinavir/ritonavir use in the TORO population to be correlated with treatment experience (number of previous antiretrovirals: 12.5 vs. 10.2 in patients with or without prior lopinavir/ritonavir experience, respectively). For those patients who had not previously received lopinavir/ritonavir, this drug would likely have been a new active agent in the patient's OB and would thus have improved the chances of virologic success. These data also further emphasizes the importance of, where possible, using enfuvirtide when a patient can still combine it with another active drug. Enfuvirtide would retain similar antiviral activity against virus resistant to other oral agents; however, across all lines of therapy, combinations of drugs from different classes have been found to be more effective than antiretroviral combinations from a single class or single agents used as monotherapy.
In conclusion, this combined analysis of the week 48 efficacy results from the 2 phase 3 studies TORO 1 and TORO 2 confirms the durable efficacy of enfuvirtide in patients previously treated with multiple antiretrovirals. Although all subgroups analyzed benefited from the addition of enfuvirtide to the OB regimen, trends observed suggest that the greatest magnitude of response when initiating enfuvirtide in combination with an OB regimen in triple-class-experienced patients can be achieved in patients with lower viral loads, higher CD4 counts, and a greater number of active antiretrovirals available to combine with enfuvirtide. Even among patients with few or no active antiretrovirals in their background regimen, however, an encouraging proportion achieved a durable virologic response. Furthermore, in patients with a PSS of 0, the mean increase in CD4 count at week 48 for patients receiving enfuvirtide was 71 cells/mm3 compared with 28 cells/mm3 for the control patients, reinforcing the argument that enfuvirtide should still be considered for patients with no active antiretrovirals in their background regimen.
Abstract
Background: The T-20 Versus Optimized Background Regimen Only (TORO) 1 and TORO 2 clinical trials are open-label, controlled, parallel-group, phase 3 studies comparing enfuvirtide plus an optimized background (OB) of antiretrovirals (n = 661) with OB alone (n = 334) in treatment-experienced HIV-1-infected patients.
Methods: The primary objective at week 48 was to investigate durability of efficacy, as measured by the percentage of patients maintaining their week 24 response or improving. Efficacy analyses used the intent-to-treat population.
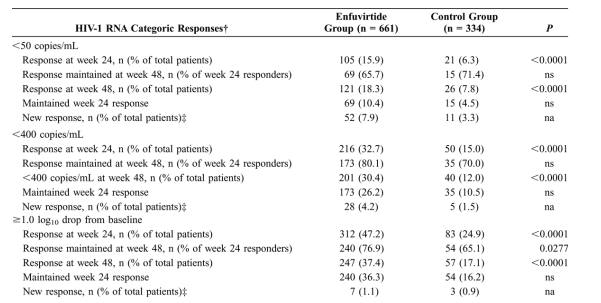
Results: A total of 73.7% of patients randomized to the enfuvirtide group remained on treatment through week 48 versus 21.3% originally randomized to the control group. At week 48, a higher proportion of week 24 responders maintained their response or were new responders in the enfuvirtide group than in the control group in each responder category: HIV-1 RNA level ≥1.0 log10 change from baseline, <400 copies/mL and <50 copies/mL (37.4%, 30.4%, and 18.3% in the enfuvirtide group vs. 17.1%, 12.0%, and 7.8% in the control group, respectively; P < 0.0001 for all comparisons). CD4 cell count increases from baseline were twice as great in the enfuvirtide group as in the control group.
Conclusion: These data demonstrate durable efficacy of enfuvirtide plus OB over 48 weeks.
RESULTS
Study Population
A total of 1013 patients were randomized during the 2 TORO studies: 673 to the enfuvirtide group and 340 to the control group. Of these, 10 patients (7 in the enfuvirtide group and 3 in the control group) withdrew their consent before receiving any study medication and 8 patients (5 in the enfuvirtide group and 3 in the control group) received study medication but had no posttreatment HIV-1 RNA measurements. Thus, the pooled ITT population comprised 661 patients in the enfuvirtide group and 334 patients in the control group. Demographic and baseline characteristics were similar between the 2 randomized treatment groups in the pooled population (Table 1).

Of the 661 patients in the enfuvirtide arm, 487 (73.7%) remained on treatment through week 48. Of the 334 in the control arm, 222 met virologic failure criteria and switched to enfuvirtide (switch group), whereas 112 did not. In the switch group, 167 (75.2%) remained on treatment through week 48. Of those not switching, 71 (63.4%) remained on treatment through week 48.
Subgroup Analyses
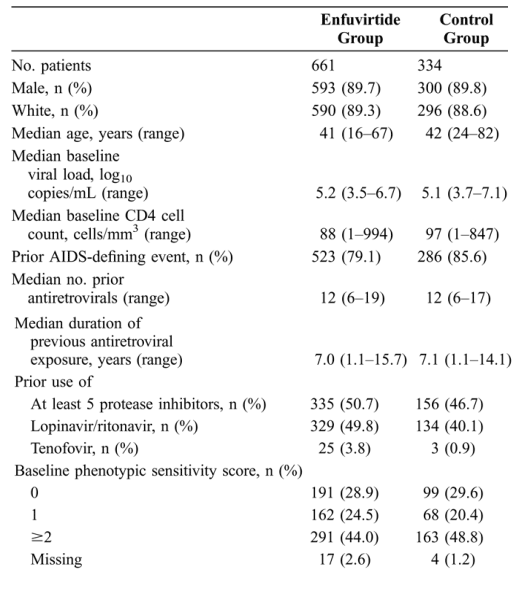
For all subgroups based on gender, race, and age (data not shown), baseline viral load levels and CD4 counts (Fig. 4), antiretroviral experience, prior use of lopinavir/ritonavir (Fig. 5), GSS or PSS (Fig. 6), the proportion of patients with a virologic response (<50 copies/mL, <400 copies/mL, ≥1 log10 drop from baseline), and the absolute increase in CD4 counts at week 48 of treatment were greater in the enfuvirtide group than in the control group.
These analyses revealed a number of trends toward greater treatment benefit for patients in certain subgroups. For both treatment groups, the percentage of patients with a virologic response was greater in subgroups with a baseline viral load of <5.0 log10 HIV-1 RNA copies/mL and a baseline CD4 count of ≥100 cells/mm3, whereas CD4 responses were greater in subgroups with a baseline CD4 count of ≥100 cells/mm3 (see Fig. 4). A clear trend toward greater treatment benefit was seen for patients with a history of treatment with fewer antiretrovirals and with no prior lopinavir/ritonavir treatment experience (see Fig. 5).
In those patients with no previous lopinavir/ritonavir experience, the use of lopinavir/ritonavir in the background regimen resulted in greater numbers of patients with a virologic response and greater mean increases in CD4 counts than was observed without the use of lopinavir/ritonavir in the background regimen (see Fig. 5A). In contrast, in patients previously treated with lopinavir/ritonavir, the use of lopinavir/ritonavir in the background regimen made no difference to the response to treatment. A treatment benefit of enfuvirtide over control was also seen, irrespective of tenofovir use (data not shown).
In addition, there was a trend toward greater benefit with a higher GSS or PSS (see Fig. 6). Of note, none of the patients in the control group with a GSS or PSS of 0 achieved a viral load of <50 HIV-1 RNA copies/mL or <400 copies/mL. In the enfuvirtide group, however, 8% of patients with a GSS of 0 and 14% of patients with a PSS of 0 achieved <400 copies/mL. Also encouraging, it was found that in patients with a PSS of 0, the mean increase in CD4 count at week 48 for patients receiving enfuvirtide was 71 cells/mm3 compared with 28 cells/mm3 for the control patients.
FIGURE 4. Virological and immunological responses at week 48 by subgroups based on baseline characteristics. A, Percentages of patients with HIV-1 RNA response at week 48 (<50 copies/mL; <400 copies/mL; ≥1 log10 drop from baseline). B, Least square mean increases in CD4 cell counts from baseline to week 48. Gray bars represent the enfuvirtide group and white bars represent the control group. All analyses were carried out on the intent-to-treat population. All comparisons between the enfuvirtide group and control group were statistically significant, P < 0.05.
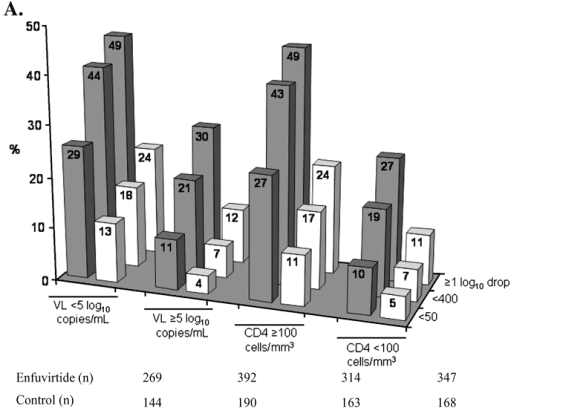
FIGURE 5. Virological and immunological responses at week 48 by subgroups based on previous treatment experience and prior use of lopinavir/r. A, Percentages of patients with HIV-1 RNA response at week 48 (<50 copies/mL; <400 copies/mL; ≥1 log10 drop from baseline). B, Least square mean increases in CD4 cell counts from baseline to week 48. Gray bars represent the enfuvirtide group and white bars represent the control group. All analyses were carried out on the intent-to-treat population. All comparisons between the enfuvirtide group and control group were statistically significant, P < 0.05 except patients reaching <50 copies/mL who had received prior LPV where P = 0.055 and patients reaching <50 copies/mL who had received ≥14 prior ARVs where P = 0.12.
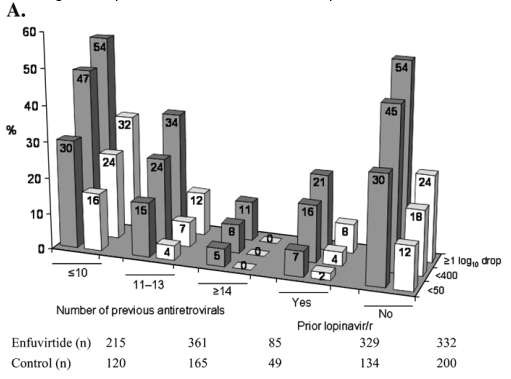
FIGURE 6. Virological and immunological responses at week 48 by subgroups based on number of active drugs in the background regimen. A, Percentages of patients with HIV-1 RNA response at week 48 (<50 copies/mL; <400 copies/mL; ≥1 log10 drop from baseline). B, Least square mean increases in CD4 cell counts from baseline to week 48. Gray bars represent the enfuvirtide group and white bars represent the control group. All analyses were carried out on the intent-to-treat population. All comparisons between the enfuvirtide group and control group were statistically significant, P < 0.05.
Durability of Response
The overall percentage of patients in each arm who maintained or improved their responder category (based on mutually exclusive categories of <50 copies/mL, ≥50 and <400 copies/mL, or ≥400 copies/mL and a decrease from baseline of ≥1.0 log10) from week 24 to week 48 was significantly higher in the enfuvirtide group compared with the control group (30.9% vs. 14.4%; P < 0.0001, X2 test). In both treatment groups, more (>65%) patients who responded within each category (<50 copies/mL, <400 copies/mL, and ≥1.0 log10 change from baseline) at week 24 maintained their response at week 48 (Table 2). In addition, there were more new responders in each category at week 48 in the enfuvirtide group than in the control group (see Table 2). These new responders represented similar proportions of the total week 48 responders in each of the treatment groups; HIV-1 RNA level <50 copies/mL in 52 (43%) of 121 patients in the enfuvirtide group and 11 (42%) of 26 patients in the control group and HIV-1 RNA level <400 copies/mL in 28 (14%) of 201 patients in the enfuvirtide group and 5 (13%) of 40 patients in the control group.
The mean decrease in HIV-1 RNA levels from baseline to week 48 was smaller in the switch group than in the enfuvirtide group (-1.1 vs. -1.9 log10 respectively, on-treatment analysis; Fig. 1). In addition, those patients who commenced enfuvirtide therapy at baseline experienced a marked drop in HIV-1 RNA levels (-1.5 log10 from baseline) within the first 2 weeks, whereas patients in the switch population exhibited a slower decrease in viral load, achieving a decrease of 1.0 log10 from baseline by week 24. The mean increase in CD4 cell count at week 48 was also smaller in the switch group (90 cells/mm3) than in the enfuvirtide group (119 cells/mm3; Fig. 2). At the time of the switch, the median viral load was 5.0 log10 and the median CD4 count was 99 cells/mm3.
Virologic Failure
In the control group, 260 patients (77.8%) met the protocol-defined criteria for virologic failure between weeks 8 and 48. In the enfuvirtide group, there were 346 (52.3%) virologic failures by week 48. The median time to virologic failure was significantly longer in the enfuvirtide group than in the control group (median: 32 weeks vs. 11 weeks; P < 0.0001; Fig. 3). Most virologic failures in both treatment groups occurred before week 24, with only a small percentage of patients (6.8% in enfuvirtide group, 7.2% in control group) experiencing virologic failure after week 24.
In the switch group, 146 patients (65.8%) met the protocol-defined criteria for virologic failure between weeks 8 and 48. Median time to failure after the switch was 13 weeks. Most failures (78.1%) had occurred by 16 weeks after the switch.
PATIENTS AND METHODS
Study Design
The TORO studies were randomized, open-label, controlled, parallel-group, phase 3 studies conducted in the United States, Canada, Mexico, and Brazil (TORO 1) and in Europe and Australia (TORO 2) as previously described.10,12 Both trials compared enfuvirtide (90 mg administered subcutaneously twice daily) plus an OB (enfuvirtide group) of 3 to 5 antiretrovirals (chosen with the aid of genotypic and phenotypic HIV resistance testing) with the OB alone (control group) over 48 weeks of treatment. Patients were HIV-1-infected adults (≥16 years old) who had been on a stable antiretroviral regimen (or no antiretroviral therapy) for longer than 4 weeks and had ≥5000 copies/mL of plasma HIV-1 RNA measured on 3 separate occasions before initiation of the trial medication. Patients were required to have at least 6 months (TORO 1) or 3 months (TORO 2) of prior experience with and/or documented resistance to at least 1 reverse transcriptase inhibitor from each of the 2 currently approved classes and at least 2 (TORO 1) or 1 (TORO 2) protease inhibitors. Patients provided written informed consent, and the protocol and patient informed consent provisions were reviewed and approved by the independent ethics committees or institutional review boards for each of the centers involved in the studies.
Patients were randomized to the enfuvirtide or control group at a 2:1 ratio. Randomization was stratified by baseline plasma HIV-1 RNA (<40,000 or ≥40,000 copies/mL) and by the use of newly approved or investigational antiretrovirals (lopinavir/ritonavir and/or tenofovir) in the OB regimen (yes or no). Changes to the treatment regimen were permitted at protocol-defined virologic failure or for toxicity management. All patients who met the criteria for failure after week 8 were allowed repeat genotypic and phenotypic resistance testing and were encouraged to modify their background regimen. After week 8, patients in the control group meeting protocol-defined virologic failure could also add enfuvirtide to a revised background regimen (switch population). The definition of virologic failure was based on 2 or 3 consecutive plasma HIV-1 RNA measures with at least 14 days between the first and the last. The 3 virologic failure criteria were a decrease of <0.5 log10 from baseline in HIV RNA level by week 8; a decrease of <1.0 log10 from baseline by week 16; or a decrease of ≥2.0 log10 from baseline, followed by a rebound of >1.0 log10 above the average of the 2 lowest values after week 6.
Genotypic and phenotypic resistance testing was carried out by ViroLogic (San Francisco, CA). For each patient, a genotypic and phenotypic sensitivity score (GSS and PSS) was calculated, representing the number of antiretrovirals in the chosen OB regimen to which the patient's virus was defined as sensitive. Genotypic sensitivity was defined using a previously published interpretation algorithm that had been adapted.10,12,17
Efficacy Parameters
The primary efficacy parameter for the TORO studies was the change from baseline in log10 plasma HIV-1 RNA level at week 24. The primary objective of the week 48 analysis was to investigate the durability of efficacy as measured by the percentage of patients who were categorized as virologic responders at week 24 and maintained or improved their virologic response at week 48. Three mutually exclusive categories of virologic responders were defined based on the plasma HIV-1 RNA load at weeks 24 and 48: <50 copies/mL, ≥50 and <400 copies/mL, or a decrease from baseline of ≥1.0 log10 but ≥400 copies/mL on 2 consecutive measures. Secondary efficacy parameters (nonmutually exclusive) included the percentage difference between enfuvirtide and control groups for patients who had viral load values <50 copies/mL, <400 copies/mL, or a ≥1.0 log10 drop from baseline at week 48; time to protocol-defined virologic failure; change from baseline to week 48 in CD4 count; and change from baseline to week 48 in viral load. Patients meeting the criteria for a particular response category at week 48 but not at week 24 were referred to as new responders.
Viral load testing was performed at every visit using the Roche Amplicor Monitor, version 5 (Roche, Palo Alto, CA). CD4 counts were assessed using centralized standard flow cytometry techniques at the second screening, baseline, every 4 weeks thereafter until week 24, and every 8 weeks thereafter until week 48.
Data Analysis
The prospectively planned week 48 analyses were carried out on data pooled from the TORO studies. This combination of data was justified, because the 2 studies had similar designs, entry criteria, and protocol-specified analyses and because the baseline demographics for each study population showed only minor differences.
All efficacy analyses were carried out on data for the intent-to-treat (ITT) population, defined as all patients who were randomized, received at least 1 dose of study medication, and had at least 1 posttreatment HIV-1 RNA measurement. Efficacy data obtained after confirmed virologic failure or after 365 days, whichever was first, were excluded from the analysis. For continuous efficacy end points, missing values were imputed for analysis using a last observation carried forward method, whereas for categoric efficacy end points, discontinuation, virologic failure, or missing values were considered failures.
Baseline viral load was the mean of the last 2 pretreatment values. The log10 HIV-1 RNA value at week 48 was defined as follows: (1) the mean of the last 2 log10 HIV-1 RNA values on completion of 48 weeks; (2) the mean of the 2 HIV-1 RNA values that confirmed virologic failure; or (3) for premature withdrawals, the mean of the last 2 log10 HIV-1 RNA values before or at withdrawal. The CD4 count at week 48 was defined from samples taken at the same time points using a single value only.
Changes from baseline to week 48 in log10 HIV-1 RNA loads and CD4 counts were analyzed by analysis of covariance, with the factors of stratum, treatment, and treatment-by-stratum interaction and the covariates of baseline PSS (for change in HIV-1 RNA loads) and baseline CD4 count (for change in CD4 counts). Virologic responder categories were analyzed by the X2 test. The median time to virologic failure was estimated by the Kaplan-Meier method. A stratified log-rank statistic was used to compare time-to-event curves between treatment groups.
Subgroup Analysis
Subgroup analyses included week 48 virologic response and change from baseline to week 48 in CD4 counts. Subgroups were based on demographic characteristics (gender, ethnicity, and age), baseline characteristics (CD4 count and plasma HIV-1 RNA level), treatment history (number of previous antiretrovirals and prior use of lopinavir/ritonavir), and potency of the background regimen (GSS and PSS [0, 1, or ≥2]). The objective of this analysis was to compare the virologic and immunologic benefit observed in the 2 treatment arms and to describe any trends that were observed in the different subgroups. P values for efficacy categoric end points were calculated using an unadjusted X2 test, whereas differences in CD4 count were analyzed by analysis of covariance with the factors of baseline CD4 count, study, treatment, and study times treatment interaction.
|
|
| |
| |
|
|
|