| |
CCR5 Deficiency Appears to Increase Risk of Developing Symptomatic West Nile Virus
|
| |
| |
CCR5 thwarts West Nile virus
Published online 17 January 2006
The Rockefeller University Press
The Journal of Experimental Medicine
A genetic mutation that protects against HIV infection increases the risk of developing clinical West Nile virus (WNV) infection, according to Glass and colleagues.
The mutation in question is a 32-bp deletion in a gene that encodes the chemokine receptor CCR5, which was identified in 1996 as a cellular coreceptor for HIV. Individuals homozygous for this mutation (CCR5Δ32) are highly resistant to HIV infection, even when repeatedly exposed to the virus. This resistance was the theoretical basis for the development of therapeutic CCR5 inhibitors, several of which are now in clinical trials. CCR5 seemed like an ideal drug target, as people missing the receptor were healthy and no diseases or infections were known to be more frequent or severe in individuals homozygous for CCR5Δ32. Mice appeared to be equally unfazed by the lack of CCR5.
But new evidence suggests that the lack of CCR5 is not completely innocuous. This group recently showed that infection with WNV--a mosquito-borne virus that has spread rapidly across the United States since 1999, often causing fatal encephalitis--was uniformly fatal in mice lacking CCR5. This finding prompted the group to look for the CCR5Δ32 allele in two cohorts of patients in the United States who had symptomatic WNV infections. They now report that 4-5% of the infected individuals were homozygous for the CCR5Δ32 allele, compared with less than 1% of the general population, suggesting that the lack of CCR5 puts people at risk for developing clinical WNV infections. The magnitude of this risk is comparable to the magnitude of protection against HIV that is conferred by this genotype. It remains to be tested whether CCR5-deficient humans, like mice, develop more severe disease because fewer protective immune cells gain access to the brain.
This study identifies not only the first genetic susceptibility factor for WNV infection but also the first association of the CCR5Δ32 allele with susceptibility to an infectious disease. These data might also raise a red flag for the use of CCR5 inhibitors in HIV-infected patients--at least in areas endemic for WNV--as such inhibitors might increase the recipients' vulnerability to severe WNV infection.
Heather L. Van Epps
Rockefeller University
CCR5 deficiency increases risk of symptomatic West Nile virus infection
"....The present study provides the first evidence that the defective CCR5 allele CCR5delta32 is a risk factor for symptomatic WNV infection, the first genetic risk factor identified for this disease....results imply that wild-type CCR5 functions as a host defense factor in WNV infection in man....Our data indicate that immunocompromised patients could be particularly susceptible to symptomatic WNV infection if CCR5 function is missing or blocked....Additional studies will be needed to address this issue, which should include close monitoring of AIDS patients treated with antagonists now in advanced stages of development targeting the HIV coreceptor activity of CCR5. Such agents are intended to imitate the homozygous CCR5delta32 genetic defect and could render AIDS patients particularly vulnerable to severe and possibly fatal WNV infection....Our results have important implications regarding the potential safety of CCR5-blocking agents now under development for the treatment of HIV/AIDS. Clinical care of individuals taking these medicines while residing in WNV-endemic areas may mandate strict measures to limit mosquito exposure and a high index of suspicion for symptoms consistent with WNV....."
Published online 17 January 2006
Journal of Experimental Medicine
William G. Glass1, David H. McDermott1, Jean K. Lim1, Sudkamon Lekhong2, Shuk Fong Yu2, William A. Frank3, John Pape4, Ronald C. Cheshier2, and Philip M. Murphy1
1 Laboratory of Molecular Immunology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892
2 Bureau of State Laboratory Services and 3 Bureau of Epidemiology and Disease Control Services, Arizona Department of Health Services, Phoenix, AZ 85007
4 Colorado Department of Public Health and Environment, Denver, CO 80246
NOTE: anywhere in this article where you see CCR532, it should read CCR5Δ32. In some cases I fixed it but in most I didn't.
ABSTRACT
West Nile virus (WNV) is a reemerging pathogen that causes fatal encephalitis in several species, including mouse and human. Recently, we showed that the chemokine receptor CCR5 is critical for survival of mice infected with WNV, acting at the level of leukocyte trafficking to the brain. To test whether this receptor is also protective in man, we determined the frequency of CCR5delta32, a defective CCR5 allele found predominantly in Caucasians, in two independent cohorts of patients, one from Arizona and the other from Colorado, who had laboratory-confirmed, symptomatic WNV infection. The distribution of CCR5delta32 in a control population of healthy United States Caucasian random blood donors was in Hardy-Weinberg equilibrium and CCR532 homozygotes represented 1.0% of the total group (n = 1,318). In contrast, CCR5delta32 homozygotes represented 4.2% of Caucasians in the Arizona cohort (odds ratios [OR] = 4.4 [95% confidence interval [CI], 1.6-11.8], P = 0.0013) and 8.3% of Caucasians in the Colorado cohort (OR = 9.1 [95% CI, 3.4-24.8], P < 0.0001). CCR5delta32 homozygosity was significantly associated with fatal outcome in the Arizona cohort (OR = 13.2 [95% CI, 1.9-89.9], P = 0.03). We conclude that CCR5 mediates resistance to symptomatic WNV infection. Because CCR5 is also the major HIV coreceptor, these findings have important implications for the safety of CCR5-blocking agents under development for HIV/AIDS.
West Nile virus (WNV), a member of the flavivirus family, is a reemerging pathogen maintained primarily between mosquitoes and birds, with humans representing one of several incidental hosts. First isolated in Uganda in 1937, WNV outbreaks have subsequently been reported in the Middle East, Africa, Western Asia, Australia, and, most recently, in the US, where it has rapidly spread westward and into Latin America and the Caribbean since arriving in New York City in 1999 (1-4).
Clinical manifestations of WNV infection can be quite variable. Epidemiologic studies indicate that although 80% of infections remain subclinical, 20% progress to a febrile illness (3). Of these, >30% of cases in the US progress to neuroinvasive disease, including meningitis, encephalitis, and/or flaccid paralysis. Survivors of severe disease typically require prolonged rehabilitation for chronic neurologic sequelae (5). To date, no specific treatments or vaccines for WNV disease have been approved by the U.S. Food and Drug Administration.
In the US, there were 16,577 laboratory-confirmed cases of WNV reported to the Centers for Disease Control and Prevention from 2001 to 2004, of which 648 were fatal (3.9%; references 6-9). Elderly and immunocompromised individuals have increased susceptibility to WNV (2, 6, 10, 11), but genetic risk factors and immunologic control mechanisms have not been fully delineated. We have recently identified the monocyte and T lymphocyte chemokine receptor CCR5 as a key factor in viral clearance and survival in a mouse model of WNV encephalitis, in which the mechanism involved trafficking of leukocytes into the infected brain (12). Consistent with this, WNV induces expression of CCR5 ligands in WNV-infected mouse brain (12, 13).
In the present study, we attempted to translate this finding to man by analyzing the distribution of the defective CCR5 allele CCR532 in two independent cohorts of symptomatic WNV-seropositive individuals from the US. CCR532 contains a 32-bp deletion within the CCR5 coding sequence and encodes a truncated protein that fails to traffic to the plasma membrane (14). CCR532 originated in Northern Europe and is found in homozygous form in 1% of Caucasians in the US (15, 16). These individuals completely lack CCR5 function, but appear healthy, suggesting that any important homeostatic functions are compensated for by other leukocyte chemoattractant receptors. In addition to regulating monocyte and T cell trafficking, CCR5 functions pathologically as a major HIV coreceptor (16). Accordingly, CCR532 homozygosity is strongly associated with resistance to HIV�\one of the strongest genetic susceptibility factors known in infectious disease (15, 17). Similarly, the hypothesis that CCR5 plays an important role in WNV pathogenesis in man predicts that the distribution of CCR532 among symptomatic patients may be strongly skewed relative to its distribution in the general population. Here, we test this prediction.
RESULTS AND DISCUSSION
Three cohorts of patients, the Colorado WNV-seropositive cohort, the Arizona WNV-seropositive cohort, and the Arizona WNV-seronegative cohort, were identified that had symptomatic illness in which WNV was considered in the differential diagnosis. Inclusion criteria for patients in these cohorts are defined in the Materials and methods section and the clinical characteristics are given in Table I. Genotypes were defined for 247 of the 261 WNV-seropositive Arizona cases (94.6%); for 148 of the 155 WNV-seropositive Colorado cases (95.5%); for 143 of 152 and 72 of 74 of the self-reporting Caucasian subsets of the Arizona and Colorado WNV-seropositive cohorts, respectively; for all WNV-seronegative random blood donors (n = 1,318); and for 145 of 154 WNV-seronegative subjects from Arizona (94.2%).
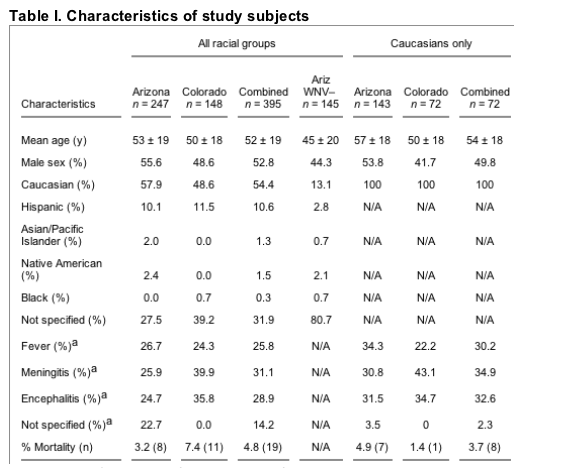
Arizona and Colorado refer to cohorts of patients with symptomatic seropositive WNV infection. Ariz WNV- refers to a second Arizona cohort of symptomatic patients in whom WNV was considered as the diagnosis but ruled out. Data pooled from the WNV+ Arizona and Colorado cohorts are also shown (Combined).
The genotype distribution observed for the combined Arizona and Colorado WNV-seropositive cohorts deviated strongly from Hardy-Weinberg equilibrium (P = 0.013), mainly because of increased frequency of CCR532 homozygotes. 17 (4.3%) of the 395 informative cases in the combined Arizona and Colorado seropositive cohorts unstratified by race were CCR532 homozygotes (Table II). This value greatly exceeds the percentage of CCR532 homozygotes reported for the general US Caucasian population (0.8-1.1%; reference 18). To quantitate the significance of this genotype-disease association, we have used as a reference the group of 1,318 healthy Caucasian random blood donors from the US, in which 1.0% were CCR532 homozygotes. Compared with this value, the increased frequency of CCR532 homozygotes in both the Arizona and Colorado WNV-seropositive cohorts was highly statistically significant (Table III). When all genotyped WNV-seropositive cases unstratified by race were compared with the US random blood donors, the odds ratios (OR) for this genetic factor were 4.7 for the Arizona cohort (95% confidence interval [CI], 2.1-10.6, P < 0.0001) and 4.2 for the Colorado cohort (95% CI, 1.6-11.3, P = 0.0018). It is important to note that this is a conservative analysis because the frequency of CCR532 homozygotes is certain to be lower in non-Caucasian racial groups from these cohorts (Table I).
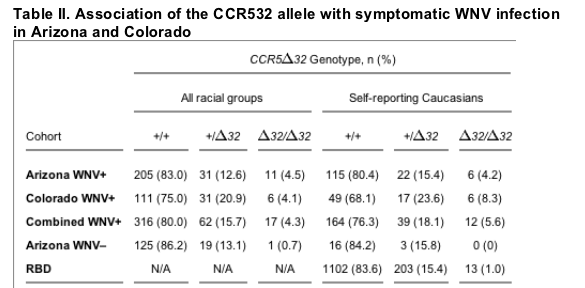
All patients from Arizona and Colorado had symptoms for which WNV infection was considered as the underlying cause. WNV+, WNV seropositive; WNV-, WNV seronegative; +, wild-type CCR5 allele; 32, CCR532 allele; RBD, random blood donors; N/A, not applicable.
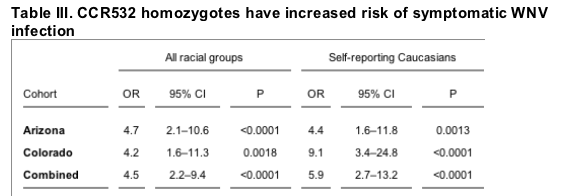
Values were calculated in comparison to the CCR532 homozygous genotype frequency in 1,318 North American Caucasian random blood donors.
Consistent with this, when the data were stratified to include only self-reporting Caucasians in both the WNV-seropositive cohorts (n = 215), the strength of the association increased by 30% (12 CCR532 homozygotes, 5.6%). When the self-reporting Caucasian portions of the two WNV-seropositive cohorts were compared separately to the random blood donor benchmark, the OR was 4.4 in the Arizona cohort (95% CI, 1.6-11.8, P = 0.0013), and 9.1 in the Colorado cohort (95% CI, 3.4-24.8, P < 0.0001).
Although unlikely, it is possible that the large increase in frequency of CCR532 homozygotes in these cohorts may be caused by an atypical geographic distribution of this allele in the general population of Arizona and Colorado. To directly address this possibility, we analyzed the distribution of CCR532 in a symptomatic Arizona WNV-seronegative cohort (n = 145). Because the CCR532 allele frequency in this cohort (7.2%) is only slightly lower than in the Arizona WNV-seropositive cohort (10.7%) and the US Caucasian random blood donors (8.7%), it is likely that this cohort contains only a slightly smaller percentage of Caucasians than the other two. Thus, given the size of the Arizona WNV-seronegative group, there is sufficient power at this allele frequency to meaningfully analyze the distribution of genotypes. Only one CCR532 homozygote was identified in the cohort (0.7%). Moreover, unlike the WNV-seropositive cohort, the observed CCR532 genotypic frequencies in the WNV-seronegative cohort did not deviate from Hardy-Weinberg equilibrium (P > 0.99). This strongly suggests that the general population in Arizona does not have an anomalous genetic structure with regard to the CCR5 locus, with a skewed increase in CCR532 homozygotes that could account for the high frequencies we observed in the WNV-seropositive cohorts. When the CCR532 homozygote genotype frequency was compared between the race-unstratified Arizona WNV-seronegative and -seropositive cohorts, an OR of 6.7 was determined (95% CI, 0.86-52.6, P = 0.07).
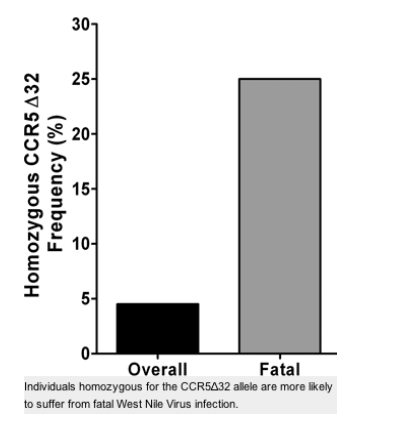
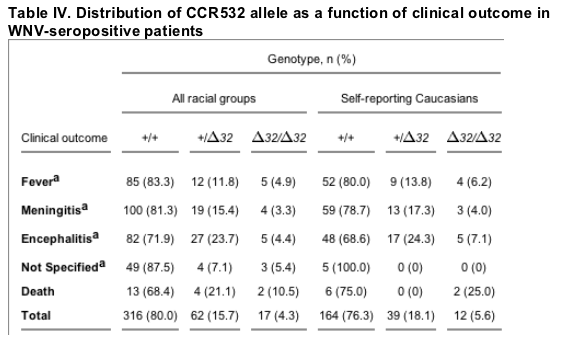
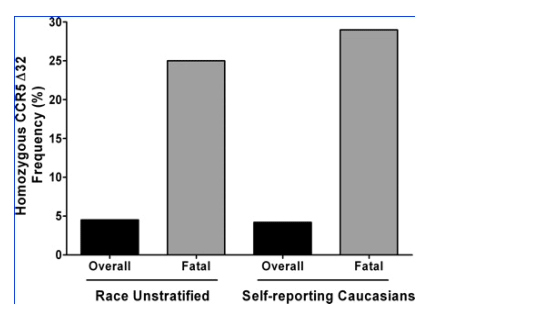
With regard to the distribution of CCR5 genotypes in symptomatic WNV-seropositive cases as a function of clinical outcome, no significant differences were observed compared with the distribution in the overall combined group (Table IV) or in the Arizona or Colorado WNV-seropositive cohorts considered separately (not depicted), with the exception of death (Fig. 1). A total of 19 of 395 individuals in the two cohorts died from WNV neuroinvasive disease (4.8%), which is similar to the national average of mortality reported for WNV cases from the past three years: 2002 (4,156 cases, 284 deaths, 6.8%); 2003 (9,862 cases, 264 deaths, 2.7%); and 2004 (2,539 cases, 100 deaths, 3.9%). The combined total for this time interval in the US is 16,577 reported cases with 648 deaths (3.9%; references 7-9). The Arizona WNV-seropositive cohort accounted for 8 of the 19 total deaths, 7 in Caucasians and 1 in a Hispanic (Table I). The Colorado WNV-seropositive cohort accounted for 11 deaths, 1 in a Caucasian. Two of the 19 fatalities (10.5%) in the combined cohorts were CCR532 homozygotes, both self-reporting Caucasians from the Arizona WNV-seropositive cohort. The ages of the two CCR532 homozygotes who died were 70 and 74, similar to the average age of the other 17 fatal cases (74 yr). The two CCR532 homozygote fatalities represent 25 and 29% of the race-unstratified (n = 8) and Caucasian (n = 7) Arizona WNV-seropositive cases who died, respectively. These values exceed the expected values based on the frequency of CCR532 homozygotes among the race unstratified and Caucasian groups in the Arizona cohort (4.5 and 4.2%, respectively), and the differences are statistically significant: OR = 8.5 (95% CI, 1.5-48.2), P = 0.04, and OR = 13.2 (95% CI, 1.9-89.9), P = 0.03, respectively (Fig. 1). This association was not observed in the Colorado cohort. When Caucasians from both cohorts were analyzed together, the association test gave an OR of 6.6 (95% CI, 1.2-37), P = 0.07.
Data were pooled from the Arizona and Colorado cohorts.
a Patients were classified into one of the four indicated disease categories by interview with the referring physician. +, wild-type CCR5 allele; 32, CCR532 allele.
Figure 1. CCR5 deficiency is associated with increased risk of death from WNV infection. Data presented are from the Arizona WNV-seropositive cohort and demonstrate that CCR532 homozygosity is associated with fatal outcome in both the race-unstratified (OR = 8.5 [95% CI, 1.5-48.2], P = 0.04) and self-reporting Caucasian (OR = 13.2 [95% CI, 1.9-89.9], P = 0.03) groups.
The present study provides the first evidence that the defective CCR5 allele CCR532 is a risk factor for symptomatic WNV infection, the first genetic risk factor identified for this disease. The association was restricted to CCR532 homozygotes and was very strong, similar in magnitude to the association we and others have previously reported for CCR532 homozygosity with the exposed, uninfected HIV resistance phenotype (e.g., OR = 6.04 [95% CI, 1.42-25.7], P = 0.02, in reference 15). Although our results are based on retrospective analysis, they are unlikely to be because of chance for six reasons. First, the results are statistically strong (an OR greater than four). Second, the results were obtained in two independent as well as geographically and temporally distinct cohorts. Third, CCR532 is a complete loss-of-function mutation and therefore homozygous individuals completely lack functional CCR5. Fourth, WNV infection is associated with infiltration of T cells and macrophages (cell types known to express CCR5) into brains of patients infected with WNV, suggesting biologically plausible involvement of CCR5 in regulating leukocyte migration to the brains of infected patients (19, 20). Fifth, WNV infection of mice induces expression of the CCR5 ligand CCL5 and accumulation of CCR5+ leukocytes in the brain (12, 13). Sixth, WNV infection in CCR5-/- mice is uniformly fatal (12).
Together these results imply that wild-type CCR5 functions as a host defense factor in WNV infection in man. CCR5 could potentially restrict WNV infection at the level of initial infection. This is not addressed by our retrospective study design and may not be feasible to resolve prospectively by measuring the association of CCR532 homozygosity with asymptomatic WNV infection because this genotype and WNV infection are both uncommon in the general population. More likely, CCR5 restricts disease progression after initial infection so that its absence results in an increased likelihood of an infected patient coming to medical attention as a symptomatic case. Our data are consistent with this interpretation, and even suggest that a fatal outcome is more likely in the absence of CCR5 (Fig. 1). Additional work will be needed to more critically address the latter point because the positive association we observed between CCR5 deficiency and death in the Arizona cohort, although statistically significant, was based on only two cases, and could not be detected in the Colorado cohort, possibly because of its smaller size.
Another limitation of our study was the incomplete information about the racial background of cases (Table I), which is needed to more precisely quantitate risk because CCR532 is primarily found in Caucasians. We have addressed this by consistently applying a conservative analysis of the data that has enabled us to test boundary conditions for quantitating the strength of the genotype-disease association. Larger prospective studies will be needed to more precisely quantitate risk as well as to further define mechanisms in man.
Our data indicate that immunocompromised patients could be particularly susceptible to symptomatic WNV infection if CCR5 function is missing or blocked. Additional studies will be needed to address this issue, which should include close monitoring of AIDS patients treated with antagonists now in advanced stages of development targeting the HIV coreceptor activity of CCR5. Such agents are intended to imitate the homozygous CCR532 genetic defect and could render AIDS patients particularly vulnerable to severe and possibly fatal WNV infection.
In summary, we have established that homozygous CCR532 is a strong host genetic risk factor for symptomatic laboratory-confirmed WNV infection, the first one identified for this disease. In contrast, the homozygous CCR532 genotype has previously been strongly associated with resistance to HIV. These genetic data imply that CCR5 plays opposite roles in HIV and WNV infection, facilitating the former and restricting the latter. Our results have important implications regarding the potential safety of CCR5-blocking agents now under development for the treatment of HIV/AIDS. Clinical care of individuals taking these medicines while residing in WNV-endemic areas may mandate strict measures to limit mosquito exposure and a high index of suspicion for symptoms consistent with WNV.
MATERIALS AND METHODS
Study populations.
The study was approved by the Office of Human Subjects Research of the US National Institutes of Health. Four patient groups were defined: (a) healthy North American Caucasian random blood donors from the National Institutes of Health Department of Transfusion Medicine (n = 1,318) previously collected under an IRB approved protocol; (b) the Arizona WNV-seropositive cohort (n = 261), defined as patients presenting with acute illness in Arizona who tested positive for WNV but negative for St. Louis encephalitis virus (SLE) by specific IgM ELISA of serum or cerebrospinal fluid (CSF) at the Arizona Department of Health Services between 5/26/04 and 10/2/04, the dates for receipt by the Department of the first and last patient samples during mosquito season in the state of Arizona in 2004; (c) the Colorado WNV-seropositive cohort (n = 155), defined as patients presenting with acute illness to a physician in Colorado who tested positive for WNV but negative for SLE by specific IgM ELISA of serum or CSF at the Colorado Department of Public Health and Environment between 7/20/03 and 9/30/03, the dates for receipt by the Department of the first and last patient samples for 2003; (d) the Arizona WNV-seronegative cohort (n = 154), defined as patients presenting with acute illness to a physician in Arizona in whom WNV and SLE had been considered in the differential diagnosis but ruled out by serological testing at the Arizona Department of Health Services between 5/26/04 and 10/2/04. In the Arizona WNV-seropositive cohort, 89% of the samples collected were available and all of these were analyzed. In the Colorado WNV-seropositive cohort, only 164 samples remained that had a sufficient volume for purification of DNA and all of these samples were analyzed. This represented 14% of all Colorado samples originally collected. The following information was requested from the medical provider during case investigation by local health department communicable disease staff: age, sex, self-reported racial group, and clinical presentation based on the Centers for Disease Control-defined clinical parameters of disease; i.e., fever, meningitis, encephalitis, and death (Table I). At the time of data collection each symptomatic patient was classified into one of the three disease categories: fever, meningitis, or encephalitis. Follow-up data on disease course and the maximal severity of symptomatic disease in each patient were not available. All fatal cases were caused by complications of WNV neuroinvasive disease. Study investigators were blinded to unique patient identifiers.
DNA isolation.
100 ml of serum or CSF was thawed for genomic DNA purification using a QiaAmp 96 DNA Blood kit according to the manufacturer's instructions (QIAGEN). Purified DNA was eluted into 100 ml of the recommended buffer and stored at 4 C until further use. DNA from random blood donors was isolated as previously described (15).
CCR532 genotyping.
Genotyping was performed by standard methods described previously (21). In brief, 2 ml of patient DNA was amplified by PCR using primers that flank the site of the 32-bp deletion: 5'-GTCTTCATTACACCTGCAGCTCTC-3' and 5'-GTCCAACCTGTTAGAGCTACTGC-3'. PCR products were analyzed by electrophoresis on a 3.0% agarose/TBE gel with known wild-type CCR5 and CCR532 amplicon controls (233 and 201 bp, respectively) and visualized with Gelstar (Cambrex) DNA staining. Each sample was tested in two independent PCR reactions, and results were concordant, as determined by two independent investigators.
Statistical analysis.
OR and 95% confidence limits were calculated using a recessive genetic model (i.e., CCR532 homozygotes compared with the combination of wild-type CCR5 and CCR532 heterozygotes) by cross-tabulation. With the exception of the death data, significance was determined by 2 tests using a two-sided P value and Yates' continuity correction, and 95% CIs were estimated using the approximation of Woolf as implemented in the Prism statistics program version 4 (GraphPad Software). Association of CCR532 homozygosity with death, due to small numbers, was analyzed by Fisher's exact test. Tests of Hardy-Weinberg equilibrium were performed using a 2 test and two degrees of freedom after using the Hardy-Weinberg equation (p2 + 2pq + q2 = 1, where p and q represent the frequency of the two alleles) to calculate expected frequencies of each of the three genotypes.
|
|
| |
| |
|
|
|