| |
Emergence of CXCR4-Using Human Immunodeficiency Virus Type 1 (HIV-1) Variants in a Minority of HIV-1-Infected Patients following Treatment with the CCR5 Antagonist Maraviroc Is from a Pretreatment CXCR4-Using Virus Reservoir
|
| |
| |
Journal of Virology, May 2006, p. 4909-4920, Vol. 80, No. 10
Mike Westby,1* Marilyn Lewis,1 Jeannette Whitcomb,2 Mike Youle,3 Anton L. Pozniak,4 Ian T. James,1 Tim M. Jenkins,1 Manos Perros,1 and Elna van der Ryst1
Pfizer Global Research and Development, Sandwich, United Kingdom,1 Monogram Biosciences, South San Francisco, California,2 Royal Free Hospital, London, United Kingdom,3 Chelsea and Westminster Hospital, London, United Kingdom4
Abstract
Antagonists of the human immunodeficiency virus type 1 (HIV-1) coreceptor, CCR5, are being developed as the first anti-HIV agents acting on a host cell target. We monitored the coreceptor tropism of circulating virus, screened at baseline for coreceptor tropism, in 64 HIV-1-infected patients who received maraviroc (MVC, UK-427,857) as monotherapy for 10 days.
Sixty-two patients harbored CCR5-tropic virus at baseline and had a posttreatment phenotype result. Circulating virus remained CCR5 tropic in 60/62 patients, 51 of whom experienced an HIV RNA reduction from baseline of >1 log10 copies/ml, indicating that CXCR4-using variants were not rapidly selected despite CCR5-specific drug pressure. In two patients, viral load declined during treatment and CXCR4-using virus was detected at day 11.
No pretreatment factor predicted the emergence of CXCR4-tropic virus during maraviroc therapy in these two patients. Phylogenetic analysis of envelope (Env) clones from pre- and posttreatment time points indicated that the CXCR4-using variants probably emerged by outgrowth of a pretreatment CXCR4-using reservoir, rather than via coreceptor switch of a CCR5-tropic clone under selection pressure from maraviroc.
Phylogenetic analysis was also performed on Env clones from a third patient harboring CXCR4-using virus prior to treatment. This patient was enrolled due to a sample labeling error. Although this patient experienced no overall reduction in viral load in response to treatment, the CCR5-tropic components of the circulating virus did appear to be suppressed while receiving maraviroc as monotherapy. Importantly, in all three patients, circulating virus reverted to predominantly CCR5 tropic following cessation of maraviroc.
Introduction
Over the past decade, numerous advances have been made in understanding the molecular mechanisms by which human immunodeficiency virus (HIV) enters CD4-positive cells. These advances have identified several potential new targets for antiviral agents. Compounds targeting viral entry have two obvious advantages over those that target the HIV-1 reverse transcriptase or protease enzymes: entry inhibitors do not depend on efficient cellular uptake or intracellular activation processes to exert their biological effects, and they are highly unlikely to show any cross-resistance with protease inhibitors or reverse transcriptase inhibitors. Viral entry has been validated as a clinically effective pathway for targeted intervention by the first fusion inhibitor, enfuvirtide (24, 25). Other classes of entry inhibitor under development target the initial binding of viral gp120 to CD4 and the interaction of gp120 with cell surface chemokine receptors that serve as coreceptors for HIV entry (CCR5 or CXCR4) (7, 33).
The HIV coreceptors represent attractive targets for drug development since they are members of the G protein-coupled receptor superfamily, a group of proteins targeted by several commonly used and well-tolerated drugs (e.g., desloratadine, ranitidine, and tegaserod) (16). CCR5 is of particular interest since a natural polymorphism exists in humans (CCR5-delta32) that leads to reduced or absent cell surface expression of CCR5 in heterozygotic or homozygotic genotypes, respectively (6). Individuals homozygotic for CCR5-delta32 appear to benefit from a natural resistance to HIV infection, while heterozygotic CCR5-delta32 is associated with reduced disease progression (6, 12, 26).
HIV-1 variants can be classified into those that exclusively use CCR5 (CCR5-tropic, or R5, viruses), those that exclusively use CXCR4 (CXCR4-tropic, or X4, viruses), and those that can use either receptor (dualtropic, or R5X4, viruses) (1). CXCR4-tropic and dualtropic viruses can collectively be termed "CXCR4 using," indicating that both viruses can infect cells using the CXCR4 as their coreceptor. Patients whose circulating virus is classified as "dualtropic" often harbor mixtures of CCR5-tropic, CXCR4-tropic, and/or dualtropic variants (37). Therefore, plasma-derived viruses that can infect CXCR4- and CCR5-expressing cells in vitro are assigned a "dual/mixed" tropism, unless subsequent clonal analysis identifies that the circulating species solely comprises dualtropic variants (5).
The genetic determinants of virus tropism appear to be concentrated within the 35-amino-acid V3 loop region of the viral envelope protein, gp120 (21). Generally, CXCR4-using viruses carry positively charged amino acids at positions 11 and/or 25 in the V3 loop, while CCR5-tropic viruses do not (8, 14). For example, results from the HOMER cohort of 1,191 patients initiating antiretroviral therapy demonstrated a strong association between a CXCR4-using phenotype and the 11/25 genotype (P < 0.0001) (3), although other analyses by the same group showed that the 11/25 genotype was not a particularly sensitive (30%) predictor of X4 phenotype (3, 4). Other algorithms exist for predicting coreceptor tropism from gp120 sequence data, including position-specific scoring matrices (PSSM) (21) and neural network approaches (35).
Almost all transmitted HIV-1 variants are CCR5 tropic; they predominate in the asymptomatic stage of infection and persist throughout the course of the disease. In contrast, CXCR4-using virus tends to emerge in the later stages of the infection in around 60% of progressing patients and its emergence coincides with an accelerated disease progression (22, 23, 32, 36). However, whether CXCR4 emergence is a cause or a consequence of severe immune system impairment is unknown (27), as little is known about the mechanisms by which CXCR4 viruses are selected during the course of infection. It is also unclear whether selective inhibition of CCR5-using strains by treatment with a CCR5 antagonist will lead to an increased rate of emergence of CXCR4 variants.
Maraviroc (MVC, UK-427,857) is an antagonist of the CCR5 coreceptor with potent and specific anti-HIV-1 activity in vitro (10). Two phase II studies have demonstrated that 10 days of maraviroc monotherapy, at doses from 100 mg to 300 mg once daily (QD) or twice daily (BID), decreased plasma viral load by >1.0 log10 copies/ml in HIV-1-infected patients (13). In these two studies, a total of 64 patients prescreened for the absence of CXCR4-using virus were treated with maraviroc for 10 days. One patient was subsequently found to harbor virus with a dualtropic phenotype at baseline and had been inadvertently enrolled because of a sample switching error at screening. The objectives of the study presented here were to monitor virus tropism in patients following maraviroc monotherapy and to characterize in detail viruses from any patients harboring CXCR4-using virus in order to determine the origins of the emergent CXCR4-using variants.
DISCUSSION
In this study, we examined the coreceptor tropism of plasma HIV-1 in 64 patients enrolled in two studies of the CCR5 antagonist maraviroc, given as short-term monotherapy to HIV-1-infected asymptomatic individuals harboring CCR5-tropic virus. Coreceptor tropism of circulating virus was assigned phenotypically before and after maraviroc treatment based on the ability of pseudoviruses, expressing gp160 envelopes amplified from patient plasma, to infect cells coexpressing high levels of CD4 and either CCR5 or CXCR4. Of the 64 patients who received maraviroc, one patient was subsequently shown to carry CXCR4-using virus at screening and was thus inadvertently enrolled in the study and one patient had no posttreatment phenotype result. Of the remaining 62 patients, CXCR4-using variants were not rapidly selected in 60, despite the counterselective pressure of treatment for 10 days with maraviroc (as evidenced by the drop in viral load of >1 log10 copies/ml experienced by 51 of these patients). CXCR4-using virus was detected at day 11 in two patients, with both patients experiencing a significant drop in viral load from baseline, indicating a clinical response to maraviroc.
Selection of CXCR4-using variants within a patient could theoretically arise by mutation from a CCR5-tropic ancestor ("coreceptor switch"). This pathway has been described in vitro using serial passage conditions in which CCR5 receptor levels are limiting (9, 17). However, other in vitro studies using a range of CCR5-tropic HIV-1 strains suggest that the molecular pathways to coreceptor switching frequently involve multiple mutations throughout the gp160 sequence, with transitional intermediates characterized as having diminished replication fitness and less efficient coreceptor usage (30). Site-directed mutagenesis studies have further revealed that accumulation of these mutations occurs in an ordered fashion, adding further complexity to this pathway of escape (28). The fact that outgrowth of preexisting variants rather than coreceptor switching was seen in these two patients treated with maraviroc is consistent with there being a high genetic barrier to this pathway.
Selection of preexisting CXCR4-using variants in patients is also theoretically possible during treatment with a CCR5 antagonist. In carefully controlled experiments, phenotypic tropism assays, such as the one used in this study, have been shown to have a sensitivity of approximately 10 to 20% (5; J. Whitcomb, unpublished observations). We did not perform a clonal analysis of plasma samples from patients who remained CCR5 tropic. However, in the majority of patients enrolled in this study, virus derived from the day 11 posttreatment plasma sample remained CCR5 tropic despite a viral load reduction of greater than 1 log10 copies/ml from baseline. Therefore, it is reasonable to conclude that any CXCR4-using variants preexisting in these patients comprised <1 to 2% of the pretreatment circulating virus.
To further evaluate whether coreceptor switching or emergence from a preexisting CXCR4-using reservoir was the most likely explanation for the change in tropism seen in patients A and B, we undertook a detailed phylogenetic analysis of individual envelope clones derived from pre- and posttreatment samples. This clonal analysis also allowed us to determine the proportions of CCR5-tropic, CXCR4-tropic, or dualtropic variants within plasma samples classified as dualtropic/mixed tropic. All the plasma virus samples assigned a dualtropic/mixed-tropic phenotype from patients A and B contained distinct CCR5-tropic and dualtropic variants. In some cases, CXCR4-tropic variants were also present. At no time point, in either of the patients, were CXCR4-using variants found to be the only circulating species, indicating that during the course of treatment there remained a CCR5-tropic component to their circulating virus. The prescreening of individual Env clones identified the existence of a minority population (2%) of CXCR4-using variants on day 1 (predose) in patient A but not in patient B. This low incidence is consistent with the CCR5-tropism assignment of this pretreatment plasma sample given the expected sensitivity of phenotypic tropism assays (5). Importantly, phylogenetic analysis of viruses present in sequential samples from these two patients indicated that the CXCR4-using variants that emerged during treatment were most likely derived from a pretreatment CXCR4-using reservoir and not by mutation from the CCR5-tropic population.
The emergence of CXCR4-using virus in patients A and B could be the result of either the outgrowth of CXCR4-using virus given a selective advantage conferred by suppression of CCR5-tropic virus or potent suppression of CCR5-tropic virus allowing the detection of the previously masked CXCR4-using virus. Both patients A and B experienced a significant reduction in viral load on treatment (0.71 log10 and 1.26 log10 copies/ml, respectively). The specific inhibition of >80% of the CCR5-tropic virus in these patients would mean that low levels of CXCR4-using virus, which previously represented a proportion of the total pretreatment virus below the limit of detection of the pseudovirus assay, could then represent a detectable proportion of the virus remaining in circulation without increasing in absolute number. While CXCR4-using viruses appear to replicate more rapidly than CCR5-tropic viruses in vitro (2, 11, 15), if this were the case in vivo, then selective treatment pressure against CCR5-tropic virus might have been expected to result in rapid domination of CXCR4 variants in the viral population of these two individuals and an earlier rebound in viral load. However, in both patients, CCR5-tropic virus persisted throughout the treatment period and became dominant after treatment was discontinued. The viral load rebound in patients A and B was also indistinguishable from that experienced by other patients in the same treatment group. These results suggest that strong selective pressure acting against the dominance of CXCR4-using viruses was preserved in these patients (4, 27, 29).
We were unable to identify retrospectively any clinical markers that would have predicted that CXCR4-using virus would emerge in patients A and B but not in the other patients receiving maraviroc or in the patients receiving placebo. Neither the baseline CD4 count (as a marker of disease progression) nor the change in viral load during therapy (which may unmask low levels of preexisting CXCR4-using virus) of patients A and B was outlying in the study group. Other factors may be predictive of the emergence of CXCR4-using viruses in patients undergoing therapy with a CCR5-specific antagonist, and it may take large epidemiology studies, such as those described recently for CCR5 and CXCR4 usage, to identify clinically useful correlates (4, 29).
In the patient with a dualtropic/mixed-tropic virus population present in pretreatment plasma (patient C), even though an overall viral load reduction was not apparent, the proportion of CCR5-tropic virus present in the viral population did decrease, demonstrating that the CCR5 tropic component of this viral population with mixed tropism remained susceptible to maraviroc. This absence of clinical response (in terms of viral load reduction) can be explained mathematically and is consistent with results in patients harboring dual/mixed tropic virus who were treated with the CXCR4 coreceptor antagonist, AMD3100 (18). Prescreening of individual Env clones derived from patient C's pretreatment plasma sample estimated that approximately 56% of the circulating virus was CCR5 tropic. Even if this CCR5-tropic virus was decreased by several log10 copies/ml, with the CXCR4-using component remaining unchanged, the maximum overall reduction in viral load would remain <0.4 log10 copies/ml (4, 31, 38).
Phenotypic characterization of individual CXCR4-using Env clones was generally consistent with the genotypic assignment of tropism, based on the changes in the V3 loop associated with CXCR4 usage (8, 14, 20, 21, 34). For example, there was 100% concordance between the PSSM method and the phenotypic assignment for CXCR4-using clones from patients A and C. However, in patient B, use of the PSSM method to assign probable tropism based on the Env sequence failed to identify the majority of CXCR4-using viruses from samples taken at follow-up time points. In these clones there were other genetic correlates of CXCR4 usage: a loss of the glycosylation site at position 6, an increase in positive charge of the V3 loop, and a change from arginine to serine at position 440 (20, 34). An algorithm that combines information from a range of sequence positions within gp120 may be needed to improve the genotypic predictability of coreceptor tropism. Indeed, regions outside the V3 loop are known to influence coreceptor tropism, as evidenced by the work of Mosier and coworkers (11, 28).
The identification of a likely recombinant virus in patient B at day 433 resolved the apparent discrepancy between the phylogenetic analysis, where the day 433 clone grouped with the CCR5-tropic clones, and the phenotypic classification of the same clone as a CXCR4-using virus. This observation indicates that the genetic determinants of coreceptor tropism were localized to the N-terminal region of the recombinant-consistent with the V3 loop playing an important role in coreceptor recognition. The origins of this recombinant clone are not certain; it could have been generated by recombination during the PCR amplification of plasma viral RNA or by a recombination event in vivo.
The possibility exists that any plasma virus sample assigned a CCR5-tropic phenotype may harbor CXCR4-using virus that could emerge when CCR5-tropic virus is suppressed. However, in patient B, prescreening large numbers of pretreatment Env clones did not predict the emergence of CXCR4-using virus posttreatment. In addition, patients A and B, who both harbored virus that was assigned a CCR5-tropic phenotype on day 1 (predose), experienced a good clinical response to treatment with maraviroc, comparable to the mean for the dose group. Therefore, it appears that CXCR4-using virus present at levels below the limit of detection of the phenotypic assay is unlikely to compromise the overall response to maraviroc, at least in the short term. Monitoring of patients treated with maraviroc in combination with an antiretroviral background in phase II/III clinical trials will help to establish the clinical relevance of these findings.
RESULTS
CXCR4-using virus is not readily selected in vivo in the majority of patients following CCR5-specific drug pressure with maraviroc. Viral tropism was assessed phenotypically (using the PhenoSense HIV Entry assay) on days 1 (predose), 11 (posttreatment), and 40 (follow-up) for all 64 patients who received maraviroc as part of trials A4001007 and A4001015, as described by Fatkenheuer et al. (13). As has already been reported, the mean decrease in viral load from baseline to nadir in these patients ranged from -0.59 log10 copies/ml (25-mg QD group) to -1.84 log10 copies/ml (300-mg BID group), with 51 patients experiencing a >1.0-log10 reduction in viral load (13). During the tropism analysis, one patient (patient C) was identified as harboring dualtropic/mixed-tropic virus pretreatment (discussed in detail below). The remaining 63 patients all harbored virus classified as exclusively CCR5 tropic at baseline. Of these, 62 patients had at least one posttreatment phenotype result (52 on days 10 and 40 and 10 on day 40 only). For one patient, neither of the posttreatment plasma samples yielded a result in the PhenoSense assay. In 60 patients, no changes in virus tropism were observed posttreatment. This indicated that CXCR4-using virus was not rapidly selected in these patients.
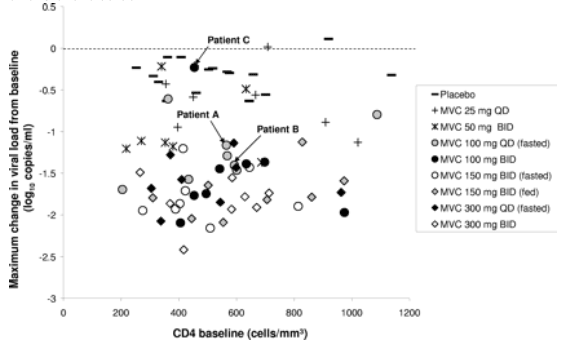
CXCR4-using virus was detected in posttreatment samples from two patients (patient A and patient B) who were previously antiretroviral treatment naive and received maraviroc as monotherapy at 100 mg QD. Neither patient A nor patient B could be distinguished from the other patients in the same treatment group in terms of baseline CD4 cell count or maximal viral load reduction (Fig. 1).
FIG. 1. Emergence of CXCR4-using virus following maraviroc treatment in 63 patients whose circulating virus was phenotypically characterized as CCR5 tropic at baseline was not related to baseline CD4 or virological response to therapy. Each symbol represents one patient. Patients A, B, and C are labeled.
Patient B was followed up and remained clinically well with no antiretroviral treatment at 1 year after the start of the study. The patient eventually started antiretroviral treatment on day 433 poststudy. At this time point, the patient's plasma viral load was not significantly different from that seen prior to dosing (4.69 log10 copies/ml at both time points) but the patient's CD4 count had declined significantly (to 219 cells/mm3 from 593 cells/mm3 on day 1 [predose]).
As highlighted above, patient C harbored mixed-tropic virus on day 1 (predose). This patient had previously received treatment with stavudine, lamivudine, didanosine, abacavir, and nevirapine in various combinations over the course of 3 years (all antiretroviral treatment ceased more than 1 year prior to enrolling in this study). Screening results suggested that this patient harbored CCR5-tropic virus only. In contrast, phenotypic characterization of virus samples obtained on days 1 (predose), 11, and 40 indicated the coexistence of viruses using CCR5 and/or CXCR4 as their entry coreceptor. Further investigation of this apparent discrepancy, by sequencing of the envelope open reading frame, revealed that the screening sample had been misidentified as belonging to patient C and instead belonged to another patient who had attended the same site on the same day. The screening sample that had been assigned to this other patient was reported as dualtropic/mixed tropic, consistent with the results for subsequent samples from patient C. After 10 days of treatment with maraviroc at 100 mg BID, patient C experienced no drop in viral load, in contrast to the other seven patients in the same treatment group (13). Pharmacokinetic parameters and CCR5 occupancy for this patient were not significantly outlying from the other patients in the group and therefore cannot account for this difference in response (not shown).
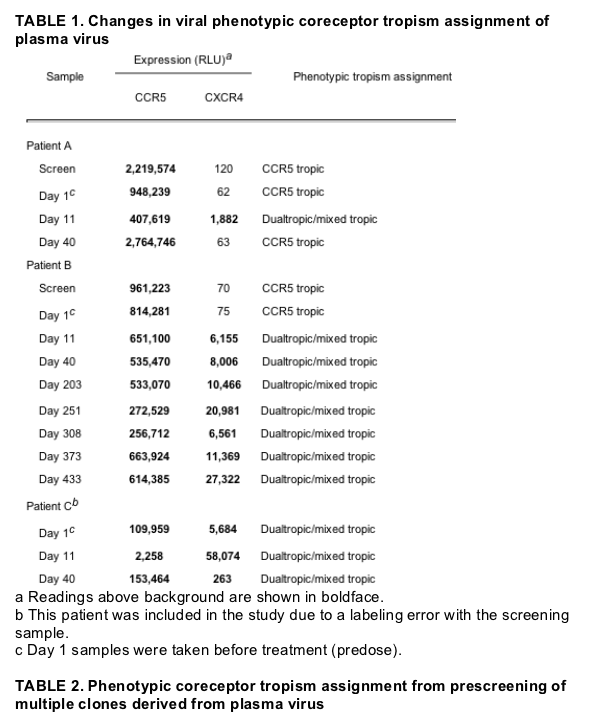
CCR5-tropic virus reemerged as the dominant circulating species after treatment with maraviroc was discontinued. For patients A and B, the phenotypic entry assay assigned a CCR5-tropic phenotype to the plasma virus from screening and day 1 samples (Table 1). In both patients, CXCR4-using virus was seen to emerge by day 11. In patient A, circulating virus reverted to a CCR5-tropic phenotype at day 40, while in patient B plasma virus remained dualtropic/mixed tropic. The RLU output suggested dominance of CCR5-tropic virus in the day 11 sample from patient A and in both day 11 and day 40 samples from patient B. However, RLU output from population phenotyping is not quantitatively related to the proportion of CCR5- or CXCR4-tropic clones in a sample. To better define the components of the viral populations in these patients, we carried out a clonal analysis of pretreatment and posttreatment samples, including follow-up samples to day 433 for patient B. In addition, the results of phenotypic prescreening of multiple individual Env clones were analyzed to assess whether a low frequency of CXCR4-using variants could be identified in pretreatment samples (Table 2). Phenotypic prescreening is routinely performed by testing 100 to 200 Env clones in individual wells of a 96-well assay plate to identify functional Env clones for inclusion in a full PhenoSense HIV Entry assay and for gp160 sequencing. Experience has shown that approximately one-third of all Env clones in the assay encode nonfunctional proteins (J. Whitcomb, unpublished observations). This prescreen information is additionally useful for getting an approximate picture of the proportion of CCR5-tropic and CXCR4-using variants from a large pool of functional Env clones. However, false positives are possible with this method if nonfunctional Env clones neighbor wells with very high RLU outputs. It is therefore important to confirm prescreening results in a PhenoSense HIV Entry assay for important clones.
For patient A, phenotypic prescreening of 97 functional Env clones at baseline identified two as CXCR4 using, both dualtropic. At day 11, although the majority of Env clones derived from this patient were CXCR4 using, a large proportion of clones remained CCR5 tropic. By day 40, only one Env clone (of 91 analyzed) from patient A appeared to be CXCR4 using. This clone did not give a signal in the full PhenoSense HIV Entry assay, nor could it be reamplified for sequencing so its tropism could not be confirmed. It is most likely that this represented a false positive in the prescreening assay and was in fact a nonfunctional envelope clone.
For patient B, no CXCR4-using virus could be identified from 185 pretreatment (screening and baseline) clones. On day 11, 40% of Env clones were assigned a CXCR4-using phenotype. This proportion progressively declined over the follow-up period, with CCR5-topic Env clones remaining the predominant species and accounting for more than 80% of viable Env clones at 1 year posttreatment.
In patient C, the circulating virus was assigned a mixed phenotype at baseline and this persisted at days 11 and 40 (Table 1). Clonal analysis revealed distinct CCR5-tropic, CXCR4-tropic, and dualtropic variants with the CCR5-tropic variants appearing to predominate (Table 2). Following 10 days of treatment with maraviroc, the proportion of viable Env clones that were CCR5 tropic had dramatically reduced, suggesting that they had been selectively inhibited by the compound in vivo. By day 40, the proportion of CCR5 clones predominated once more, indicating that short-term monotherapy with maraviroc in this patient had not resulted in a permanent shift in overall coreceptor tropism.
CXCR4-using variants are genetically distinct from CCR5-tropic clones in each patient, indicating a separate ancestral origin. Full gp160 sequences were obtained for 12 Env clones per time point amplified from pretreatment, day 11, and posttreatment samples for each patient. Alignment of these sequences to reference consensus sequences representing the various HIV-1 group M genetic subtypes indicated that the circulating viruses from all three patients were typical of group M subtype B strains (data not shown). A phylogenetic analysis of the Env clones was then performed for each patient, with each clone being phenotypically classified as CCR5 tropic or CXCR4 using based on its properties in the PhenoSense HIV Entry assay. As shown in Fig. 2, CXCR4-using variants detected in each patient clustered together in the neighbor-joining trees and were genetically distinct from CCR5-tropic variants. In patient A, the CCR5-tropic viruses clustered on a number of small branches, with the main branch bifurcating: one subbranch representing a small outlying group of CCR5-tropic variants supported by a very high bootstrap value and the other subbranch representing the group of CXCR4-using variants (Fig. 2A). Within the subbranch of CXCR4-using variants, the gp160 sequences were monophyletic, with the day 11 virus clustering with the day 1 (predose) virus supported by bootstrap values in the neighbor-joining tree. This was also seen in the maximum parsimony and maximum likelihood trees (not shown). This indicates that the CXCR4-using virus detected at day 11 had emerged from a pretreatment reservoir of circulating CXCR4-using virus and had not evolved by mutation of a CCR5-tropic clone during maraviroc treatment.
The phylogenetic tree assembled from gp160 clones isolated from patient B contained sequences obtained during follow-up of this patient over the course of one year postenrollment. The tree comprised three branches: a main branch, which included CCR5-tropic viruses and CXCR4-using viruses and an outlying group of CCR5-tropic viruses (Fig. 2B). The CXCR4-using viruses formed a monophyletic group with a bootstrap value of 88% and were distinct from the CCR5-tropic virus. This suggested that they had arisen from an unidentified ancestor and not any identified CCR5-tropic virus. This topology was also obtained using the maximum parsimony and maximum likelihood methods (data not shown). A single outlying CXCR4-using clone was identified in the day 433 sample (Fig. 2B). A detailed analysis to understand its likely origin is discussed below.
As discussed earlier, patient C had circulating CCR5-tropic and CXCR4-using viruses on day 1 (predose). This can clearly be seen in the phylogenetic tree as two distinct branches representing, separately, CCR5-tropic and CXCR4-using variants (Fig. 2C). The significant bootstrap values observed in the neighbor-joining tree were supported in the maximum likelihood and maximum parsimony trees (not shown). This confirmed that the CXCR4-using virus sequenced at the posttreatment time points had evolved from the preexisting circulating CXCR4-using virus and not from the CCR5-tropic population.
A single Env clone from patient A was assigned a CCR5-tropic phenotype and clustered with the CXCR4-using clones (Fig. 2A). Closer examination of the tropism data obtained with the pseudovirus derived from this Env clone revealed low RLU readings on CCR5-expressing cells in the PhenoSense HIV Entry assay (1,206 RLU), indicating that it had low infectivity. This may explain why a signal was not obtained on the CXCR4-expressing cells. Consistent with this was the fact that the sequence of the V3 loop for this clone was indicative of a CXCR4-using clone (Table 3).
V3 loop sequence analysis of Env clones from patients A, B, and C identifies genetic correlates of coreceptor tropism. For all three patients, V3 loop sequences of clones assigned a CXCR4-using phenotype differed from the sequences of clones assigned a CCR5-tropic phenotype (Table 3). Consistent with published algorithms for predicting coreceptor use from V3 loop sequence (8, 14, 20, 21), the predominant change was the presence of a basic amino acid at position 11 or 25 (11/25 algorithm) of the CXCR4-using clone. Use of this method to predict CXCR4 coreceptor usage correctly assigned all CXCR4-tropic/dualtropic clones from patients A and C and 10 of the 13 unique V3 loop sequences of CXCR4-tropic/dualtropic clones from patient B (Table 3). All 10 CXCR4-using clones from patient A and 6 of 28 CXCR4-using clones from patient B carried an arginine at position 11; similarly, all 20 CXCR4-using clones from patient C carried a lysine at position 25. In general, basic substitutions in the V3 loop were more frequent in the dualtropic and CXCR4-tropic clones. For example, in patient C an increase in charge of the V3 loop was observed caused by the insertion of two amino acid residues (arginine and tryptophan) between positions 17 and 18.
Genotypic prediction of viral tropism based on the PSSM method of Jensen et al. (21) correlated with the phenotypic tropism assigned to the virus for all clones from patient A and 11/12 unique V3 loop sequences of Env clones from patient C. However, in patient B, the PSSM scores failed to predict CXCR4 usage in all but three sequenced clones assigned a CXCR4-tropic phenotype. Instead, the loss of the g15 N-linked glycosylation site (amino acids 6 to 8), reported to enable more efficient use of CXCR4 (34) and observed in all 28 CXCR4-using clones, appeared to be the most reliable genotypic predictor of CXCR4 usage in this patient.
Identification of a genetically outlying CXCR4-using variant generated by recombination between CCR5-tropic and CXCR4-using ancestors. As identified above, one genetically outlying CXCR4-using clone was identified from the day 433 sample of patient B that grouped with the CCR5-tropic clones in the phylogenetic tree. Its position in the tree varied dramatically, depending upon whether N-terminal or C-terminal regions of the gp160 gene were excluded from the analysis, suggesting that it may represent a recombinant variant. The Env sequence from this outlying CXCR4-using clone was compared to the sequences of the CCR5-tropic and CXCR4-using variants from the same patient that were closest in similarity, using the RIP tool from the Los Alamos HIV Sequence Database. This analysis tool is typically used to identify circulating recombinant forms in patients with evidence of infection with mixed genetic subtypes. The output of the RIP analysis clearly shows a likely recombination event between a CXCR4-using virus (in the N-terminal region of Env) and a CCR5-tropic variant (in the C-terminal region of Env) (Fig. 3). The likely crossover falls at approximately amino acid residue 350 (1,050 bp into Env), corresponding to the C4 domain within gp120. To confirm this finding, two phylogenetic trees were drawn, using the 36 Env clones from the last three time points (data not shown). In the first tree, in which N-terminal domains up to and including the V3 loop were included in the analysis, the outlying CXCR4-using clone from day 433 strongly associated with the CXCR4-using clones. In contrast, in the second tree, in which the C-terminal domains were included from C4 to the end of gp41, the outlying CXCR4-using clone from day 433 associated with the CCR5-tropic clones. No other recombinant viruses were identified among the 107 other Env clones analyzed from this patient over the course of approximately 1 year of follow-up, suggesting this was a relatively rare event.
MATERIALS AND METHODS
Clinical trial design. Trials A4001007 and A4001015 were designed to evaluate the effect of short-term monotherapy with maraviroc on viral load and to assess its safety and tolerability in HIV-positive patients. Eighty patients were prescreened for the presence of CCR5-tropic virus before being treated with either placebo or maraviroc as monotherapy for a period of 10 days. Maraviroc was administered at doses ranging from 25 mg QD to 300 mg BID. The primary efficacy endpoint was the change in viral load from baseline to day 11. Patients were followed up for 30 days posttreatment. The trial designs and clinical results are described in detail elsewhere (13).
Coreceptor tropism analysis. Circulating virus was tested from the plasma of patients using the PhenoSense HIV Entry assay for coreceptor tropism (Monogram Biosciences Inc., South San Francisco, CA) (5). The entire HIV envelope coding sequence was amplified from plasma samples by PCR and was ligated into a pCXAS expression vector to create an envelope expression vector library. Virus particles carrying envelope glycoproteins derived from the plasma virus were produced by transfecting HEK293 cells with the purified envelope expression vector library and an HIV-1 genomic vector lacking the envelope-encoding region and containing a firefly luciferase gene. The ability of the pseudoviruses to infect U87 cells expressing CD4 and either CCR5 or CXCR4 was assessed by measuring luciferase relative light units (RLU). Plasma virus was assigned a particular tropism if infection of cells expressing the relevant coreceptor resulted in an RLU reading above the background. Tropism was confirmed by the inhibition of viral replication in each cell type by a specific coreceptor inhibitor.
For the clonal analysis of plasma-derived virus, approximately 100 to 200 individual Env clones were prescreened for viability and tropism in single-well cultures of CCR5- or CXCR4-expressing cells. At each time point, 12 viable clones were selected for confirmation of the prescreening phenotypic tropism assignment and Env sequencing for genotypic tropism assignment and phylogenetic analysis. Sequencing was carried out using Big Dye chain terminator chemistry (PE Biosystems, Foster City, CA) and the ABI PRISM 3700 DNA analyzer. Sequence electropherograms were aligned and edited using customized software (Gene Codes Corp., Ann Arbor, MI). Protein sequences were aligned using the Clustal W program (39), and the alignment was edited using the GeneDoc program (http://www.psc.edu/biomed/genedoc) to obtain the V3 loop alignment.
V3 loop sequences of clones were also analyzed to predict CXCR4 coreceptor usage on the basis of the PSSM (http://ubik.microbiol.washington.edu/computing/pssm/) (21), the loss of the g15 N-linked glycosylation site (amino acids 6 to 8) (34), and the presence of basic amino acid residues at positions 11 and/or 25 (8, 14, 20). The results of these genotypic analyses were compared to the coreceptor tropism assigned to the individual Env clones from the phenotypic assay described above.
Phylogenetic analysis. Phylogenetic analysis of Env clones was carried out using nucleotide sequence alignments of near-full-length gp160. Neighbor-joining trees were generated using the Clustal W program, and bootstrapped values for a 1,000 repetitions were obtained (19). The options to disregard gapped columns and to attempt to correct for multiple hits were selected in the Clustal W tree program. Two short hypervariable regions in V4 and V5 were also excluded from the phylogenetic analysis (corresponding to HXB2 nucleotide positions 402 to 410 and 462 to 465): these regions are rich in insertion/deletion mutations, and experience has shown that they are often therefore associated with convergent evolution. A consensus sequence was generated from the earliest set of clones and used as an outgroup to root the tree. Viral subtyping was performed by alignment of the gp160 nucleotide sequences from the patient viruses against the HIV-1 subtype reference sequences available from the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov/content/hiv-db/mainpage.html). For all trees shown in this study, individual clones were assigned a coreceptor tropism based on their behavior in the phenotypic assay.
Further phylogenetic analyses were performed on sequences from each patient, using the HXB2 reference sequence as an outgroup to root the tree and a sequence from each of the other patients as representatives of current circulating subtype B virus. These alignments were edited to exclude any gapped regions, and phylogenetic trees were constructed using the maximum likelihood method and maximum parsimony from PHYLIP 3.65 (http://evolution.genetics.washington.edu/phylip.html): SEQBOOT was used to provide bootstrap values for input into the maximum parsimony program, and a single tree was generated from the output using CONSENSE.
Identification of circulating recombinant forms. Consensus gp160 sequences were assembled from individual clusters of CCR5-tropic (five clusters) and CXCR4-using (three clusters) Env clones, based on the output from the phylogenetic analysis. Outlying Env clones were then queried against these clusters using the RIP tool available at the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov/content/hiv-db/mainpage.html). Phylogenetic trees were then constructed using partial gp160 sequences on either side of any points of recombination identified to identify the genetic origins of the recombinant virus.
References
1. Berger, E. A., R. W. Doms, E. M. Fenyo, B. T. Korber, D. R. Littman, J. P. Moore, Q. J. Sattentau, H. Schuitemaker, J. Sodroski, and R. A. Weiss. 1998. A new classification for HIV-1. Nature 391:240.[CrossRef][Medline]
2. Bleul, C. C., L. Wu, J. A. Hoxie, T. A. Springer, and C. R. Mackay. 1997. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 94:1925-1930.[Abstract/Free Full Text]
3. Brumme, Z. L., W. W. Dong, B. Yip, B. Wynhoven, N. G. Hoffman, R. Swanstrom, M. A. Jensen, J. I. Mullins, R. S. Hogg, J. S. Montaner, and P. R. Harrigan. 2004. Clinical and immunological impact of HIV envelope V3 sequence variation after starting initial triple antiretroviral therapy. AIDS 18:F1-F9.[CrossRef][Medline]
4. Brumme, Z. L., J. Goodrich, H. B. Mayer, C. J. Brumme, B. M. Henrick, B. Wynhoven, J. J. Asselin, P. K. Cheung, R. S. Hogg, J. S. Montaner, and P. R. Harrigan. 2005. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J. Infect. Dis. 192:466-474.[CrossRef][Medline]
5. Coakley, E., C. J. Petropoulos, and J. M. Whitcomb. 2005. Assessing chemokine co-receptor usage in HIV. Curr. Opin. Infect. Dis. 18:9-15.[Medline]
6. Dean, M., M. Carrington, C. Winkler, G. A. Huttley, M. W. Smith, R. Allikmets, J. J. Goedert, S. P. Buchbinder, E. Vittinghoff, E. Gomperts, S. Donfield, D. Vlahov, R. Kaslow, A. Saah, C. Rinaldo, R. Detels, and S. J. O'Brien. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273:1856-1862.[Abstract/Free Full Text]
7. De Clercq, E. 2002. New anti-HIV agents and targets. Med. Res. Rev. 22:531-565.[CrossRef][Medline]
8. De Jong, J. J., A. De Ronde, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66:6777-6780.[Abstract/Free Full Text]
9. Dejucq, N., G. Simmons, and P. R. Clapham. 2000. T-cell line adaptation of human immunodeficiency virus type 1 strain SF162: effects on envelope, vpu and macrophage-tropism. J. Gen. Virol. 81:2899-2904.[Abstract/Free Full Text]
10. Dorr, P., M. Westby, S. Dobbs, P. Griffin, B. Irvine, M. Macartney, J. Mori, G. Rickett, C. Smith-Burchnell, C. Napier, R. Webster, D. Amour, D. Price, B. Stammen, A. Wood, and M. Perros. 2005. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49:4721-4732.[Abstract/Free Full Text]
11. Douek, D. C., L. J. Picker, and R. A. Koup. 2003. T cell dynamics in HIV-1 infection. Annu. Rev. Immunol. 21:265-304.[CrossRef][Medline]
12. Eugen-Olsen, J., A. K. Iversen, P. Garred, U. Koppelhus, C. Pedersen, T. L. Benfield, A. M. Sorensen, T. Katzenstein, E. Dickmeiss, J. Gerstoft, P. Skinhoj, A. Svejgaard, J. O. Nielsen, and B. Hofmann. 1997. Heterozygosity for a deletion in the CKR-5 gene leads to prolonged AIDS-free survival and slower CD4 T-cell decline in a cohort of HIV-seropositive individuals. AIDS 11:305-310.[CrossRef][Medline]
13. Fatkenheuer, G., A. L. Pozniak, M. A. Johnson, A. Plettenberg, S. Staszewski, A. I. M. Hoepelman, M. S. Saag, F. D. Goebel, J. K. Rockstroh, B. J. Dezube, T. M. Jenkins, C. Medhurst, J. F. Sullivan, C. Ridgeway, S. Abel, I. T. James, M. Youle, and E. van der Ryst. 2005. Efficacy of short-term monotherapy with maraviroc, a novel CCR5 co-receptor antagonist in HIV-infected patients. Nat. Med. 11:1170-1172.[CrossRef][Medline]
14. Fouchier, R. A. M., M. Brouwer, S. M. Broersen, and H. Schuitemaker. 1995. Simple determination of human immunodeficiency virus type 1 syncytium-inducing V3 genotype by PCR. J. Clin. Microbiol. 33:906-911.[Abstract]
15. Grivel, J.-C., M. L. Penn, D. A. Eckstein, B. Schramm, R. F. Speck, N. W. Abbey, B. Herndier, L. Margolis, and M. A. Goldsmith. 2000. Human immunodeficiency virus type 1 coreceptor preferences determine target T-cell depletion and cellular tropism in human lymphoid tissue. J. Virol. 74:5347-5351.[Abstract/Free Full Text]
16. Gurrath, M. 2001. Peptide-binding G protein-coupled receptors: new opportunities for drug design. Curr. Med. Chem. 8:1605-1648.[Medline]
17. Harrowe, G., and C. Cheng-Mayer. 1995. Amino acid substitutions in the V3 loop are responsible for adaptation to growth in transformed T-cell lines of a primary human immunodeficiency virus type 1. Virology 210:490-494.[CrossRef][Medline]
18. Hendrix, C. W., C. Flexner, R. T. MacFarland, C. Giandomenico, E. J. Fuchs, E. Redpath, G. Bridger, and G. W. Henson. 2000. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob. Agents Chemother. 44:1667-1673.[Abstract/Free Full Text]
19. Higgins, D. G., J. D. Thompson, and T. J. Gibson. 1996. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 266:383-402.[Medline]
20. Hoffman, N. G., F. Seillier-Moiseiwitsch, J. Ahn, J. M. Walker, and R. Swanstrom. 2002. Variability in the human immunodeficiency virus type 1 gp120 Env protein linked to phenotype-associated changes in the V3 loop. J. Virol. 76:3852-3864.[Abstract/Free Full Text]
21. Jensen, M. A., F.-S. Li, A. B. van't Wout, D. C. Nickle, D. Shriner, H.-X. He, S. McLaughlin, R. Shankarappa, J. B. Margolick, and J. I. Mullins. 2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J. Virol. 77:13376-13388.[Abstract/Free Full Text]
22. Koot, M., I. P. Keet, A. H. Vos, R. E. de Goede, M. T. Roos, R. A. Coutinho, F. Miedema, P. T. Schellekens, and M. Tersmette. 1993. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 118:681-688.[Abstract/Free Full Text]
23. Koot, M., R. van Leeuwen, R. E. de Goede, I. P. Keet, S. Danner, J. K. Eeftinck Schattenkerk, P. Reiss, M. Tersmette, J. M. Lange, and H. Schuitemaker. 1999. Conversion rate towards a syncytium-inducing (SI) phenotype during different stages of human immunodeficiency virus type 1 infection and prognostic value of SI phenotype for survival after AIDS diagnosis. J. Infect. Dis. 179:254-258.[CrossRef][Medline]
24. Lalezari, J. P., K. Henry, M. O'Hearn, J. S. Montaner, P. J. Piliero, B. Trottier, S. Walmsley, C. Cohen, D. R. Kuritzkes, J. J. Eron, Jr., J. Chung, R. DeMasi, L. Donatacci, C. Drobnes, J. Delehanty, and M. Salgo. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 348:2175-2185.[Abstract/Free Full Text]
25. Lazzarin, A., B. Clotet, D. Cooper, J. Reynes, K. Arasteh, M. Nelson, C. Katlama, H. J. Stellbrink, J. F. Delfraissy, J. Lange, L. Huson, R. DeMasi, C. Wat, J. Delehanty, C. Drobnes, and M. Salgo. 2003. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N. Engl. J. Med. 348:2186-2195.[Abstract/Free Full Text]
26. Meyer, L., M. Magierowska, J. B. Hubert, C. Rouzioux, C. Deveau, F. Sanson, P. Debre, J. F. Delfraissy, and I. Theodorou. 1997. Early protective effect of CCR-5 32 heterozygosity on HIV-1 disease progression: relationship with viral load. AIDS 11:F73-F78.[CrossRef][Medline]
27. Moore, J. P., S. G. Kitchen, P. Pugach, and J. A. Zack. 2004. The CCR5 and CXCR4 coreceptors-central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retrovir. 20:111-126.[CrossRef][Medline]
28. Mosier, D. E., A. Ramos, R. Nedellec, R. Offord, and O. Hartley. 2005. Differential impacts of V1-V2 and V3 mutations on resistance to a CCR5 inhibitor. Antivir. Ther. 10:S67.
29. Moyle, G. J., A. Wildfire, S. Mandalia, H. Mayer, J. Goodrich, J. Whitcomb, and B. G. Gazzard. 2005. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J. Infect. Dis. 191:866-872.[CrossRef][Medline]
30. Pastore, C., A. Ramos, and D. E. Mosier. 2004. Intrinsic obstacles to human immunodeficiency virus type 1 coreceptor switching. J. Virol. 78:7565-7574.[Abstract/Free Full Text]
31. Philpott, S., B. Weiser, K. Anastos, C. M. Kitchen, E. Robison, W. A. Meyer III, H. S. Sacks, U. Mathur-Wagh, C. Brunner, and H. Burger. 2001. Preferential suppression of CXCR4-specific strains of HIV-1 by antiviral therapy. J. Clin. Investig. 107:431-438.[Abstract/Free Full Text]
32. Philpott, S. M. 2003. HIV-1 coreceptor usage, transmission, and disease progression. Curr. HIV Res. 1:217-227.[CrossRef][Medline]
33. Pierson, T. C., R. W. Doms, and S. Pohlmann. 2004. Prospects of HIV-1 entry inhibitors as novel therapeutics. Rev. Med. Virol. 14:255-270.[CrossRef][Medline]
34. Polzer, S., M. T. Dittmar, H. Schmitz, and M. Schreiber. 2002. The N-linked glycan g15 within the V3 loop of the HIV-1 external glycoprotein gp120 affects coreceptor usage, cellular tropism, and neutralization. Virology 304:70-80.[CrossRef][Medline]
35. Resch, W., N. Hoffman, and R. Swanstrom. 2001. Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology 288:51-62.[CrossRef][Medline]
36. Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X.-L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489-10502.[Abstract/Free Full Text]
37. Singh, A., and R. G. Collman. 2000. Heterogeneous spectrum of coreceptor usage among variants within a dualtropic human immunodeficiency virus type 1 primary-isolate quasispecies. J. Virol. 74:10229-10235.[Abstract/Free Full Text]
38. Skrabal, K., V. Trouplin, B. Labrosse, V. Obry, F. Damond, A. J. Hance, F. Clavel, and F. Mammano. 2003. Impact of antiretroviral treatment on the tropism of HIV-1 plasma virus populations. AIDS 17:809-814.[CrossRef][Medline]
39. Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680.[Abstract/Free Full Text]
|
|
| |
| |
|
|
|