| |
Detection of Low-Level NNRTI Drug Resistance Using a Highly Sensitive Genotypic Resistance Test, may be associated with therapy failure
|
| |
| |
"Selection and persistence of non-nucleoside reverse transcriptase inhibitor-resistant HIV-1 in patients starting and stopping non-nucleoside therapy" using a sensitive genotypic resistance test to detect low level NNRTI (K103N) mutations
AIDS: Volume 20(5) 21 March 2006 p 701-710
Palmer, Saraha; Boltz, Valeriea; Maldarelli, Franka; Kearney, Marya; Halvas, Elias Kd; Rock, Dianeb; Falloon, Judithc; Davey, Richard T Jrc; Dewar, Robin Le; Metcalf, Julia Ab; Mellors, John Wd; Coffin, John Ma
From the aHIV Drug Resistance Program, National Cancer Institute, National Institutes of Health, Frederick
bNational Institute of Allergy and Infectious Diseases /CCMD Clinic
cLaboratory of Immunoregulation of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland
dDivision of Infectious Diseases, University of Pittsburgh, Pittsburgh, Pennsylvania
eSAIC, Frederick, Maryland.
Standard genotyping and phenotyping methods provide a composite of the HIV sequences present, or of their phenotypic properties, but can only detect variants constituting a major fraction of the virus population, typically > 25%, and are not quantitative [25-28]. More sensitive detection methods for minor HIV-1 populations are required [29,30]. This study addressed this by developing an allele-specific, RT-initiated polymerase chain reaction (PCR) technique for quantifying NNRTI-resistant variants encoding 103N (AAT or AAC alleles) at low frequencies (0.1%). The method was then used to study the appearance and decay of these variants in patients starting and stopping NNRTI therapy.
"....To our knowledge, this is the first report of measurable levels of preexisting 103N variants in drug-naive patients.... Minor populations of drug-resistant HIV-1 variants that arise during virus replication before initiation of antiretroviral therapy may contribute to treatment failure [34,35]. It has not been possible to test this because assays for such minor variants were not available. Therefore, the allele-specific real-time RT-PCR assay described here was developed..... Recent reports showed that multidrug-resistant viruses acquired during primary HIV-1 infection (primary drug resistance) can persist indefinitely in the absence of antiretroviral therapy [38-41]...... The appearance and decay of K103N variants were studied in six patients before, during, and after NNRTI-containing therapy. Four of the six patients showed persistence of the 103N variants after cessation of NNRTI therapy..... Two standard genotyping assays during this interval detected only wild-type virus...Our results indicate that acquired drug resistance can also persist for more than 5 years after cessation of therapy and may not be detected by standard genotyping. In the most extreme case (Fig. 2d), mutant virus remained at nearly 100% almost 6 years after cessation of efavirenz treatment...... The clinical significance of the persistent variants is not defined, but preliminary evidence suggests that minor drug-resistant variants that are missed by standard genotyping [27] can lead to failure of subsequent treatment regimens.... the method described here will help to define the clinical significance of low-frequency drug-resistant HIV-1 variants in treatment-naive and treatment-experienced patients...."
Abstract
Background: Understanding the selection and decay of drug-resistant HIV-1 variants is important for designing optimal antiretroviral therapy.
Objective: To develop a high-throughput, real-time reverse transcriptase (RT) polymerase chain reaction (PCR) assay to quantify non-nucleoside reverse transcriptase inhibitor (NNRTI)-resistant variants K103N (AAT or AAC alleles) at frequencies as low as 0.1%, and to apply this to monitor these variants before, during, and after NNRTI therapy.
Methods: HIV-1 RNA in longitudinal plasma samples obtained from patients starting and stopping NNRTI therapy was converted to cDNA and the target sequence region amplified and quantified by real-time PCR. Approximately 107 copies/reaction provided a template for a second round of PCR using primers that discriminated between the mutant and wild-type alleles. Amplification specificity was confirmed by thermal denaturation analysis.
Results:
Frequencies of 103N similar to assay background (0.029%) were observed in longitudinal samples from 9 of 12 treatment-naive patients;
three patients had transient increases in 103N frequency to a range of 0.21-0.48%, which was 7-16.5 times assay background.
Analysis of longitudinal plasma samples from six NNRTI-experienced patients showed three patterns: persistence of 103N variants after stopping NNRTI therapy, codon switching of 103N between AAC and AAT during NNRTI therapy, and decay of 103N variants to below assay background after cessation of NNRTI therapy.
Conclusions: Allele-specific RT-PCR quantified the emergence and decay of drug-resistant variants in patients over a broad range of frequencies (0.1-100%). The rate of decay of K103N variants after stopping NNRTI therapy was highly variable.
Introduction
Although antiretroviral therapy has reduced the morbidity and mortality associated with HIV-1, development of drug resistance remains a major obstacle to long-term drug efficacy. Minor populations of drug-resistant HIV-1 variants existing before therapy have been hypothesized to be an important cause of treatment failure [1]. Another major source of drug-resistant variants is their selection during antiretroviral therapy [2,3]. Following removal of the selection pressure, either by cessation of therapy or by transmission of the virus to an untreated individual, variants encoding drug resistance often decline to undetectable levels in the virus population, albeit at variable rates [4,5]. Such drug-resistant variants can rapidly reappear following reinitiation of the antiretroviral therapy but their role in treatment failure has not been clearly established [6,7]. Determining the clinical significance of these minor drug-resistant populations requires accurate detection and quantification.
Resistance to non-nucleoside reverse transcriptase inhibitors (NNRTI), such as nevirapine or efavirenz, is conferred by single point mutations in the reverse transcriptase (RT)-coding region of the pol gene. A common NNRTI resistance mutation is lysine to asparagine at residue 103 (K103N), which causes broad cross-resistance to the NNRTI class [8-13]. The 103N mutation appears to have little impact on the replicative capacity of HIV-1, allowing 103N variants to persist as dominant species after cessation of NNRTI therapy [14]. Long-term persistence of NNRTI-resistant variants is potentially important in patients who stop and restart NNRTI-containing regimens or receive single-dose nevirapine for the prevention of mother-to-child transmission of HIV-1 [15-23]. A recent report suggested that single-dose nevirapine can decrease the efficacy of a subsequent nevirapine-containing treatment regimen even though nevirapine-resistant variants are not detected postpartum by standard genotyping methods [24]. This observation raises the possibility that minor populations of nevirapine-resistant variants are responsible for the reduced efficacy.
Standard genotyping and phenotyping methods provide a composite of the HIV sequences present, or of their phenotypic properties, but can only detect variants constituting a major fraction of the virus population, typically > 25%, and are not quantitative [25-28]. More sensitive detection methods for minor HIV-1 populations are required [29,30]. This study addressed this by developing an allele-specific, RT-initiated polymerase chain reaction (PCR) technique for quantifying NNRTI-resistant variants encoding 103N (AAT or AAC alleles) at low frequencies (0.1%). The method was then used to study the appearance and decay of these variants in patients starting and stopping NNRTI therapy.
AUTHOR DISCUSSION
Minor populations of drug-resistant HIV-1 variants that arise during virus replication before initiation of antiretroviral therapy may contribute to treatment failure [34,35]. It has not been possible to test this because assays for such minor variants were not available. Therefore, the allele-specific real-time RT-PCR assay described here was developed. Blind testing of virus mixtures showed that the assay accurately quantified the 103N variant from 100% to 0.01% with an assay background of 0.006%. This background level is similar to the single-step mutation rate and may reflect mutations arising during reverse transcription or first-round PCR amplification. Analysis of assay reproducibility showed that the mean coefficient of variation was 31% across the range of mutant frequencies. The concordance between real-time RT-initiated PCR and single-genome sequencing in five clinical samples across a range of mutant frequencies further validated the assay.
To determine the frequency of preexisting K103N variants, plasma samples were analyzed from 12 drug-naive patients. Most of the samples had mutant frequencies statistically indistinguishable from assay background, implying a steady-state level of K103N less than approximately 0.03%. In four samples, however, the frequency of 103N (AAC) was 7-16.5 times higher than the assay background in repeated assays. To our knowledge, this is the first report of measurable levels of preexisting 103N variants in drug-naive patients. Further improvement in assay sensitivity will be needed to detect preexisting variants in most drug-naive individuals.
A possible concern with PCR-based assays is their vulnerability to genetic variation in the region complementary to the allele-specific primers [30,36,37]. Indeed, we found that certain mutations outside of codon 103, but within the selective primer region, can affect the efficiency of amplification of wild-type and mutant alleles. In extreme, but rare, cases, it is necessary to redesign one or more primers based on the consensus sequence of the patient's virus (unpublished data). In addition, we have readily adapted the assay to HIV-1 subtype C virus, allowing analysis of most samples from a South African cohort (S. Palmer et al., unpublished data). We have also adapted the assay to detect other common NNRTI mutations, including Y181C and G190A (unpublished data).
The selection and persistence of drug-resistant HIV-1 variants are the principal obstacles to successful long-term therapy. Recent reports showed that multidrug-resistant viruses acquired during primary HIV-1 infection (primary drug resistance) can persist indefinitely in the absence of antiretroviral therapy [38-41]. Our results indicate that acquired drug resistance can also persist for more than 5 years after cessation of therapy and may not be detected by standard genotyping. In the most extreme case (Fig. 2d), mutant virus remained at nearly 100% almost 6 years after cessation of efavirenz treatment. The failure of wild-type virus to reappear may reflect the fact that it is reduced to very low levels by a long (greater than 3 year) period of NNRTI therapy. In addition, the relatively low selective disadvantage of 103N variants [14] and stochastic evolution could lead to delayed reappearance of wild-type virus [34].
The clinical significance of the persistent variants is not defined, but preliminary evidence suggests that minor drug-resistant variants that are missed by standard genotyping [27] can lead to failure of subsequent treatment regimens [42-44]. In addition, minor K103N variants may explain why HIV-infected women who have received single-dose nevirapine to prevent mother-to-child transmission are less likely to have virological suppression with subsequent nevirapine-containing regimens [16,43,45]. Indeed, follow-up studies of women who have received single-dose nevirapine show that the K103N and Y181C variants detected with our assay can persist for at least 1 year after these variants are no longer detected by standard genotype [22,46].
In four of the six patients studied, the K103N mutant was mostly encoded by AAC; the AAT codon was present at approximately 100-fold lower levels, with similar proportionality between the codons during and after treatment. Since the proteins encoded by these two variants are identical, the different frequencies of the two codons probably reflects differences in the mutation rates of AAA to AAC or AAT, leading to variation in preexisting levels of the two mutants. Selective forces at the level of RNA structure or codon preference may also be involved. By contrast, two of the patients showed switching of the preponderant codon for 103N from AAC to AAT and back to AAC. In one of the patients, single-genome sequencing revealed that the switch from AAC to AAT (99%) was likely related to its linkage to M184V, which selected by lamivudine therapy [27]. However, other mechanisms may also be involved.
In conclusion, the method described here will help to define the clinical significance of low-frequency drug-resistant HIV-1 variants in treatment-naive and treatment-experienced patients. Additional studies of the frequency and variation of preexisting or persistent drug-resistant variants using this method should provide further insights into mechanisms of evolution of drug resistance.
Results
Analysis of a blinded test panel of mutant/wild-type viral mixtures
To evaluate the accuracy and precision of the assay for the 103N (AAC) allele, a blinded panel of mutant/wild-type virus mixtures was tested (Table 1). The 103N allele was detected down to a mutant frequency of 10-4 (0.01%). The mean assay background for 100% wild-type virus was 0.006%. The coefficient of variation for the replicates was 31% for samples containing mutant virus and 40% for the sample containing no mutant. A linear regression analysis of the expected percentage of 103N versus the mean of the four runs revealed a slope of 0.92, not statistically different from 1.0 (P = 0.13). A larger set of analyses of wild-type virus from cell culture gave a 103N (AAC) background of 0.029% (SD, 0.022) (Fig. 1a). A conservative accuracy for the assay was considered to be to levels of mutant as low as 0.1%; assay values below this level but above background were interpreted as indeterminate. For quality control, any sample yielding a coefficient of variation of greater than 100% was rejected or reanalyzed.
Comparison of assay performance
The use of single-genome sequencing to detect and quantify individual viral variants encoding drug resistance in virus populations has been described previously [27]. To compare allele-specific PCR assay with single-genome sequencing, the percentage of wild-type and mutant alleles at codon 103 was measured by both assays in samples from two NNRTI-experienced patients, patients 1 (Fig. 2a) and 5 (Fig. 2e), as described below. These samples were selected because the levels of one or both mutants, as detected by allele-specific PCR, were adequate for quantification by single-genome sequencing (Table 2). For patient 1, similar percentages of the wild-type and AAC mutant alleles were detected by allele-specific and single-genome sequencing assays in the three samples analyzed. Both assays also gave concordant results for the two samples from patient 5, detecting approximately equal frequencies of the AAC and AAT mutants in a background of 3-7% wild type.
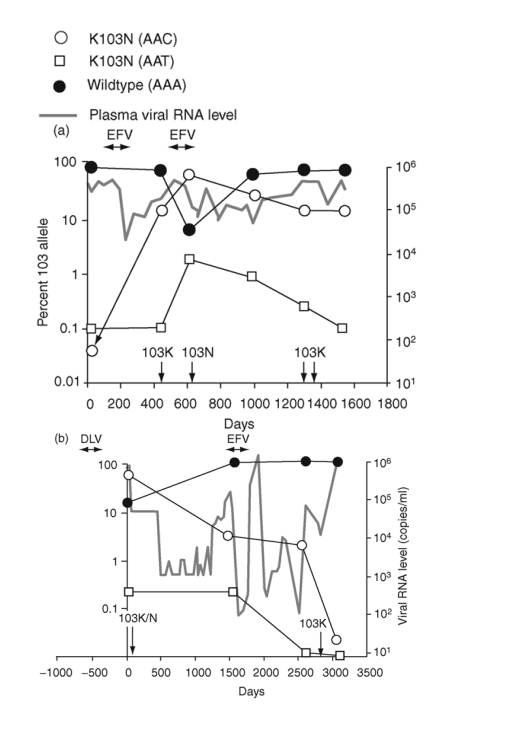
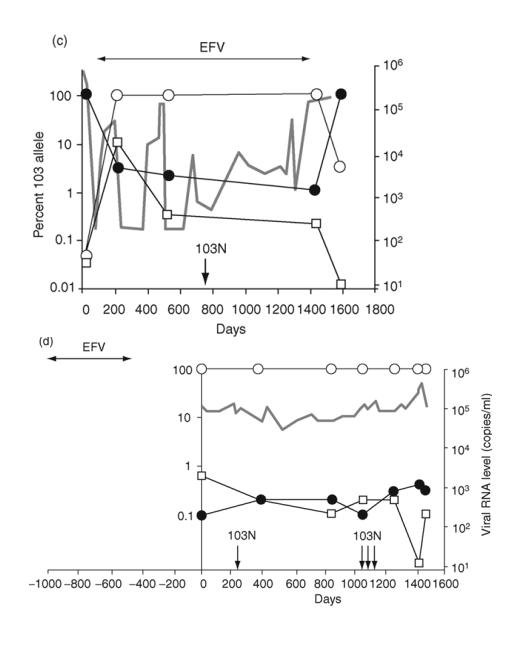
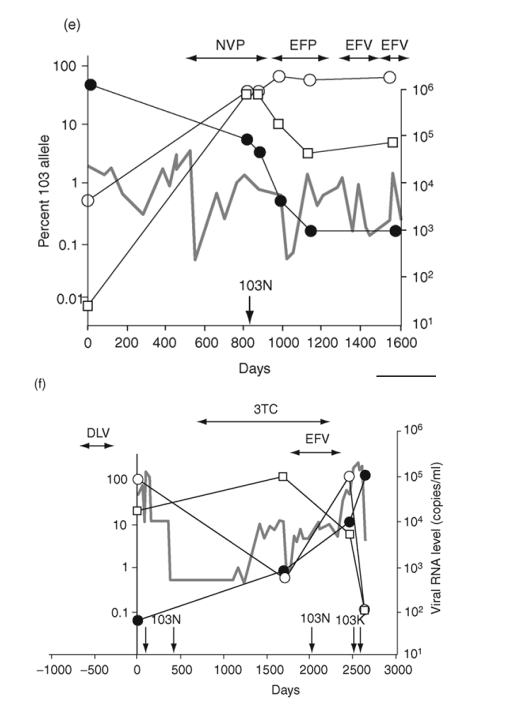
Fig. 2. Emergence and persistence of non-nucleoside reverse transcriptase inhibitor (NNRTI)-resistant variants (a-d) and codon switching between AAC and AAT mutant variants (e and f) in patients starting and stopping NNRTI-containing regimens. Longitudinal plasma samples were obtained from patients before and during NNRTI therapy and after its cessation. Patients were also treated with various combinations of other antiretroviral drugs as well as interleukin 2 during the course of the study (not shown). The horizontal arrows indicate the periods of NNRTI treatment. Vertical arrows identify the timing and results of standard genotype analyses. EFV, efavirenz; DLV, delavirdine; NVP, nevirapine; 3TC, lamivudine.
Frequency of 103N variants in untreated patients
To study the pretherapy frequency of K103N variants [1,33], plasma samples were assayed from 12 treatment-naive patients; three of these patients had longitudinal samples tested over 1 year or more. Repeated measurements (n = 24) of plasmid-derived control wild-type virus (103K; AAA) established the assay background to be 0.029% (SD, 0.022) (Fig. 1a). Overall, the percentage of mutant virus in most patients (9 of 12) was indistinguishable from the assay background (Fig. 1b), with an average of 0.032% (SD, 0.027) (P = 0.42 using multilevel random effects model). Four samples from three patients (indicated by * in Fig. 1b) exhibited transient increases in K103N to 0.21%, 0.38%, 0.41%, and 0.48%, respectively, or 7-16.5 times the assay background (Fig. 1b). Repeat extraction and analyses of these samples gave consistent results (not shown). Each of these four values exceed the 95% prediction interval for the 87 data points of 0.186% and so are unlikely to have occurred by chance. Therefore, the frequency of 103N variants in the NNRTI-naive patients was usually near or below assay background, although transient increases could occur.
Emergence and persistence of NNRTI-resistant variants in patients
The appearance and decay of K103N variants were studied in six patients before, during, and after NNRTI-containing therapy (Fig. 2). Four of the six patients showed persistence of the 103N variants after cessation of NNRTI therapy (Fig. 2a-d). For the first patient (Fig. 2a), the frequency of AAC 103N variant increased from pretherapy baseline of 0.03% to > 90% following two cycles of exposure and failure of an efavirenz-containing regimen. After cessation of efavirenz, the 103N variants declined gradually but persisted at approximately 15% for at least 2.5 years. Two standard genotyping assays during this interval detected only wild-type virus [27].
A second patient (Fig. 2b) was treated with delavirdine monotherapy 1 year prior to sampling and received efavirenz-containing therapy during the sampling interval. Over the 1500 days before receiving efavirenz, the frequency of 103N (AAC) variants declined from nearly 100% to 5%, then stabilized after efavirenz exposure for several years, before declining to background levels over an additional 2.5 years.
The third patient was treated with an efavirenz-containing regimen for almost 4 years (Fig. 2c). After 200 days, 103N (AAC) variants made up nearly 100% of the population and remained at this level until efavirenz was discontinued. The AAC variants then declined rapidly, although they still formed 3% of the virus population 6 months later.
The fourth patient was treated with an efavirenz-containing regimen for about 1.5 years and then efavirenz was stopped approximately 1 year before the first sample (Fig. 2d). In contrast to the decline in 103N variants observed in the prior three patients after stopping efavirenz, AAC variants persisted at high levels for at least 5 years after efavirenz was discontinued.
To test whether the persistence of 103N variants after cessation of efavirenz resulted from linkage of 103N to mutations conferring resistance to other antiretroviral drugs that were continued, sequences from 19-25 genomes were obtained by single-genome sequencing of three plasma samples from the patient described in Fig. 2a. The results (Table 3) showed that sequences with either 103K or 103N were polymorphic for resistance alleles at nearby sites, indicating that linkage of resistance mutations was not responsible for persistence of 103N variants.
Mutations from AAA to AAC or AAT can change codon 103 from K to N. AAC variants usually predominate, but AAT variants can often be detected. Two patients showed a pattern of switching between the 103N codons (Fig. 2e,f). In one, both variants rose from near background level to about 50% during 6 months of therapy with a nevirapine-containing regimen (Fig. 2e). After the switch to an efavirenz-containing regimen, the AAT codon declined to 2% and the AAC codon increased to approximately 90%.
In the second patient with codon switching, two switches occurred during the study (Fig. 2f). This patient was treated with delavirdine monotherapy 1 year prior to sampling and received efavirenz-containing therapy for 660 days near the end of sampling. Following delavirdine monotherapy, AAC variants made up 83% of the population and AAT variants made up 17%. At the next sampling time (nearly 5 years later), AAC variants had declined to about 1%, being replaced by AAT variants (99%). By the end of efavirenz-containing therapy, a second codon switch had occurred, with AAC variants predominating (85%) and AAT variants becoming minor (5%). Analysis of the virus populations by single-genome sequencing (Table 4) showed that the first codon switch to AAT was associated with linkage to an M184V mutation selected by concomitant lamivudine therapy.
Methods
Viruses
Infectious plasmid clones of HIV-1LAI encoding 103K (AAA) or 103N (AAC) were prepared as previously described [31].
Virus stocks were produced from plasmid clones as described [32]. Defined virus mixtures at varying ratios were prepared in HIV-seronegative human plasma. The mixtures were stored at -80 C and tested as a blinded panel.
Clinical specimens
All clinical samples were collected from participants attending the NIAID/CCMD Clinic at the National Institutes of Health, Bethesda, Maryland. The treatment-naive patients included in this study met the following criteria: age 18 years or older, minimum of 8 weeks since first known HIV-1 seropositivity, no history of opportunistic infection or AIDS-related malignancy, plasma HIV-1 RNA > 1000 copies/ml, and no prior exposure to antiretroviral drugs. The six treatment-experienced patients studied were selected based on a history of virological failure on a NNRTI-containing regimen, at least one standard genotyping that detected K103N in plasma virus, and the availability of longitudinal samples after the discontinuation of NNRTI therapy. All participants provided written informed consent. Testing of samples was approved by the Institutional Review Board of NIAID, National Institutes of Health.
Preparation of HIV RNA standards for viral RNA quantification
A 1400 bp fragment from the Xba I to the Xma1 sites of HIV-1LAI [31] was ligated into the transcription vector pTri-Amp-19 (Ambion; Austin, Texas, USA). Plasmids were purified on miniprep columns (Qiagen; Valencia, California, USA) and linearized using EcoRI. The resulting DNA template was then transcribed with T7 RNA polymerase using the Ambion Megascript kit. RNA was quantified spectrophotometrically, diluted to 108 copies/ml, and stored at -80 C. Serially diluted wild-type HIV-1 transcripts were used in the first round PCR (described below) to generate a standard curve for quantification of viral RNA in plasma.
Preparation of DNA standards for second round PCR amplification
To prepare DNA standards, a region of HIV-1 RT from bases 2195 to 2818 of the HIV-1 genome was amplified from wild-type and mutant transcripts by using primers 2195F (AAACAATGGCCATTGACAGAAGA) and 2818R (CCAAAGGAATGGAGGTTCTTTCTG). The DNA product, encompassing codons 22 to 229 of RT, was purified on gels, quantified, diluted to 108 copies/ml, divided into aliquots, and stored at -80 C. Serially diluted DNA was used to generate a standard curve for quantifying the amount of wild-type or mutant sequence in each sample.
Viral RNA extraction and first round amplification
Between 250 and 1800 ml plasma (containing ≥ 10 000 copies HIV-1 RNA) was diluted to 1800 ml with Tris-buffered saline and centrifuged at 16 000 X g for 1 h at 4 C. After centrifugation, the virion pellet was treated with 50 ml of 5 mmol/l Tris-HCL (pH 8.0) containing 200 mg proteinase K for 30 min at 55 C, then with 200 ml of 5.8 mol/l guanidinium isothiocyanate containing 200 mg glycogen. Isopropanol (270 ml) was added and the lysate was centrifuged at 16 000 X g for 30 min. The pellet was washed with 70% ethanol and resuspended in 30 ml of 5 mmol/l Tris-HCL (pH 8.0) containing 1 mmol/l dithiothreitol and 31 U RNase inhibitor (RNase out; Invitrogen; Carlsbad, California, USA). The PCR was commenced with 1 ml of each deoxynucleoside trisphosphate (dNTP; 12.5 mmol/l each) and 1.6 ml primers 2195F and 2818R (10 mmol/l); these were added to 10 ml extracted viral RNA (in three separate wells), no-template controls, and diluted RNA transcripts (3 X 106 to 300 copies/10 ml), followed by denaturation at 65 C for 5 min. For first round, real-time RT-PCR with SYBR green detection, 25.8 ml reverse transcription Quantitect Sybr Green RT-PCR mix (Qiagen) containing 20 ml master mix (1X), 0.4 ml RT mix, and 5.4 ml RNase-free water was added to the denatured viral RNA-primer-dNTP mixture (final volume 40 ml). Amplifications, data acquisition, and analysis were performed using an Opticon DNA engine fluorescence detector (Biorad; Hercules, California, USA) for one cycle of 50 C for 40 min, one cycle of 95 C for 15 min, and 50 cycles of 95 C for 15 s, 51 C for 30 s, and 69 C for 1 min, followed by reading of the plates at 73 C (approximately 2 C below the product melting temperature and above that of non-specific products). To confirm specificity of amplifications, PCR products were subjected to thermal denaturation analysis from 70 C to 80 C. Samples that did not yield the correct melting temperature for the specific amplification product were censored. HIV-1 RNA in test samples were estimated by comparing the sample cycle threshold with that of known concentrations of RNA transcripts. In most cases, this value agreed well with results of commercial HIV-1 RNA assays. Controls lacking RNA templates were run to exclude contamination with PCR product.
Second round allele-specific amplification
Approximately 107 copies of amplification product from the first round RT-PCR were used in a second round of real-time PCR, with primers that discriminated between the mutant and wild-type sequences as well as non-specific primers to quantify total template. In each case, first round PCR products were diluted in 5 mmol/l Tris-HCL (pH 8.0) and 5 ml portions of this were added to the second round real-time PCR with SYBR green quantification. To quantify total viral sequences, reaction conditions were (25 ml final volume) 1X SYBR Green buffer (SYBR Green PCR Core Reagents, Applied Biosystems; Foster City, California, USA), 250 mmol/l each of the four dNTPs, 4 mmol/l MgCl2, 300 nmol/l of each primer 2280F (5'-TTGGGCCTGAAAATCCATACAAT-3') and 2470R (5'-ATCACCCACATCCAGTACTGTTACTGA-3'), and 2 U Amplitaq Gold polymerase (Applied Biosystems). For allele-specific amplification of the 103N (AAC or AAT) mutants, conditions were identical except the reverse primer was replaced by 2465R(AAC) (5'-CCACATCCAGTACTGTTACTGATTGG-3') or 2466R(AAT) (5'-CCCACATCCAGTACTGTTACTGATTCA-3'). Samples were evaluated by real-time PCR using the following conditions: 95 C for 12 min, and 45 cycles of 95 C for 15 s, 61.5 C for 20 s, 69 C for 1 min, and 74 C for 20 s, followed by reading of the plates. PCR products were subjected to thermal denaturation analysis as above. To quantify the sequences with the wild-type K103 allele, similar conditions were used except for use of the K103 reverse primer 2468R(AAA) (5'-CCCACATCCAGTACTGTTACTGATTCT-3'). The quantity of mutant or wild-type sequences was estimated by comparison of the sample cycle threshold values with those from dilutions of amplified DNA derived from wild-type or mutant transcripts, as appropriate. The percentage of sequences containing 103N was calculated by dividing the quantity of mutant sequences by the quantity of total sequences and multiplying by 100. To allow for polymorphism in the samples that might affect amplification of all species equally, values were further normalized to the sum of all species detected. Non-specific and allele-specific amplifications were performed simultaneously. All sample reactions were performed in triplicate, standards in duplicate, and two background controls were included in each run (100% K103 or 103N plasmid as appropriate). Additional controls lacking DNA templates were carried out for both non-selective and allele-specific reactions to exclude contamination.
|
|
| |
| |
|
|
|