| |
Single-center experience of darunavir (DRV) in 38 HIV-infected adults enrolled in US expanded access program
|
| |
| |
Reported by Jules Levin
HIV-DART, Frontiers in Drug Development for Antiretroviral Therapies, 10-14 December 2006, Cancun, Mexico.
J Gathe, Jr., MD1; K Degazon, MD1; C Mayberry, PA1; J Nemecek, MD1; R Yeh, PharmD2; A Perniciaro, BA3; and D Israel, PharmD3
1Therapeutic Concepts, Houston, TX, USA; 2Tibotec Therapeutics, Bridgewater, NJ, USA, and University of Houston, Houston, TX,USA; 3Tibotec Therapeutics, Bridgewater, NJ, USA
Abstract
Background: Darunavir (DRV; TMC114 [PREZISTA]), co-administered with ritonavir (DRV/r), is a protease inhibitor (PI) recently approved in the US, Canada, and Russia to treat HIV infection in antiretroviral (ARV)-experienced adults. A multi-center, international expanded access program (EAP; study
TMC114-C226) opened October 2005 for ARV-experienced patients failing multiple regimens.We present our single-center experience from the DRV EAP.
Methods: Enrollment at our site began November 2005. The primary objective was to provide early access to DRV/r for ARV-experienced HIV-1-infected patients who have failed multiple ARV regimens and have limited treatment options. The secondary objective was to gather additional safety information on DRV/r. Eligible patients were ≥18 years, PI- (2 prior regimens), NRTI- and NNRTI-experienced, with limited or no treatment options. Patients with stable HBV and/or HCV were allowed. CD4 count ≦200 cells/mm3 restriction was lifted in January 2006. Patients received DRV/r 600/100mg bid plus other ARVs. (No PI combinations with DRV/r were allowed, except for atazanavir and indinavir.)
Results:
At the time of analysis (October 13, 2006), 38 and 23 patients, respectively, had completed 12 weeks and 24 weeks (±1 week) of therapy, as well as all protocol-required visits.
Mean age was 48 years, with 92% male; 61% were white and 5% were co-infected with HCV. At baseline, mean viral load (VL) and median CD4 count were 126,775 copies/mL and 144 cells/mm3, respectively.
Mean number of prior ARVs was 11, and 95% of patients previously used ≥3 PIs. Overall, 37% of patients had previously used tipranavir and 39% enfuvirtide (ENF).
On study, 23 patients (61%) received ENF; 15 (39%) were ENF-naive.
Through Week 24, 9 adverse events (AEs) (2 rashes [mild], 1 nausea [mild], 2 increased ALT [Grade 2], 1 hypercholesterolemia [Grade 2], 1 hypertriglyceridemia [Grade 3], 1 increased creatinine [Grade 2], and 1 increased total bilirubin [Grade 2]) were considered possibly related to DRV. Two serious AEs, unrelated to DRV, were reported (ENF injection-related hematoma requiring surgery and one death).
After 12 weeks, 54% of patients achieved HIV RNA <50 copies/mL. At Week 24, 50% of patients had <50 copies/mL; 91% had achieved this by Week 12. Mean VL reduction at Week 24 was -2.17 log10 copies/mL and mean CD4 increase was 109 cells/mm3. Of patients with CD4 data at baseline and Week 24 (n=22), 10 had CD4 <50 cells/mm3 at baseline. By Week 24, 80% of that group had CD4 >100 cells/mm3.
Conclusion: DRV/r 600/100mg bid demonstrated clinically meaningful
changes in VL and CD4 counts in our population of treatment-experienced
patients. Most patients achieved undetectable VL within the first 12 weeks
of therapy with responses maintained throughWeek 24. DRV/r was well
tolerated. AEs were infrequent, with no unexpected events observed beyond
those reported in the POWER studies.
Introduction
Darunavir (DRV; TMC114 [PREZISTA]) is a protease inhibitor (PI) that exhibits significant in vitro antiviral activity against wild-type virus and multi-drug resistant HIV-1 strains.1
DRV is currently approved in the US, Canada, Russia, and Argentina with low-dose ritonavir at a recommended dose of 600/100mg bid for the treatment of HIV infection in antiretroviral treatment-experienced adult patients.
In the Phase IIb, randomized, controlled, multi-center POWER 1 and 2 studies, DRV/r 600/100mg bid demonstrated significant virologic and immunologic efficacy in treatment-experienced patients after 24 and 48 weeks of treatment. DRV/r was also shown to be generally safe and well tolerated.
Based on the efficacy and safety data from the POWER studies, a multi-center, international expanded access program (EAP) was established in October 2005 for HIV-1 infected patients who had failed multiple antiretroviral (ARV) regimens and had limited treatment options.
We present here interim data on DRV/r in 38 patients from one of the participating EAP centers.
Methods
The primary objective of the EAP was to provide early access to DRV/r for highly ARV-experienced patients who failed multiple ARV regimens.
The secondary objective was to accumulate additional data on the safety and tolerability of DRV in combination with low-dose ritonavir and other ARVs.
Eligibility criteria
Males and females >18 years of age with:
- Documented HIV-1 infection and limited or no treatment options due to virological failure or intolerance to multiple ARV regimens.
- Three-class experience, including 2 prior PI-based regimens, or evidence of resistance to three or more classes.
- Incomplete virologic suppression on current regimen and risk of clinical or immunologic progression.
- CD4 count ≦200 cells/mm3 (restriction was lifted in January 2006).
Patients co-infected with clinically stable chronic hepatitis B or C were allowed.
All patients provided informed written consent.
Study design
Eligible patients were to continue DRV/r until treatment-limiting toxicity, pregnancy, loss to follow-up, discontinuation from the study, or commercial availability of DRV.
Patients were instructed to return for visits at 4 and 12 weeks following initiation of DRV/r and every 12 weeks thereafter.
Use of PIs other than atazanavir or indinavir was not allowed.
Use of investigational medications within the last 30 days was prohibited.
Adverse events (AEs) leading to treatment interruption or discontinuation, and all serious AEs, with the exception of most non-fatal AIDS-defining illnesses, were collected at each visit.
Physicians were advised to follow current treatment guidelines for changing ARV treatment regimens due to virologic failure.
Data on CD4 count and VL were collected at each study visit and measured locally as per standard of care.
Virologic response was summarized by the proportion (%) of patients with HIV RNA <50 copies/mL and/or ≥1.0 log10 reduction in HIV RNA from baseline.
Analyses were based on all treated patients using observed cases.
Results
As of the data cutoff date of October 26, 2006, 38 and 22 patients, respectively, had completed 12 and 24 weeks of therapy and all protocol-required visits, and had CD4 and VL measurements available.
Baseline demographics and patient characteristics are shown in Table 1. Nearly all (92%) patients were men, and nearly 40% of patients were non-white.
The mean number of prior ARVs was 11, with 95% of patients having used ≥3 PIs previously.
- Tipranavir was previously used in 37% of patients and enfuvirtide (ENF) in 39%.

During the study, 23 (61%) patients received ENF, 15 of whom were ENF-naive.
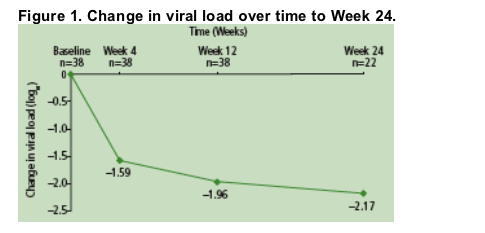
Mean change in plasma VL from baseline was -1.96 log10 copies/ml at Week 12 (n=38) and -2.17 log10 copies/mL (n=22) at Week 24 Figure 1.
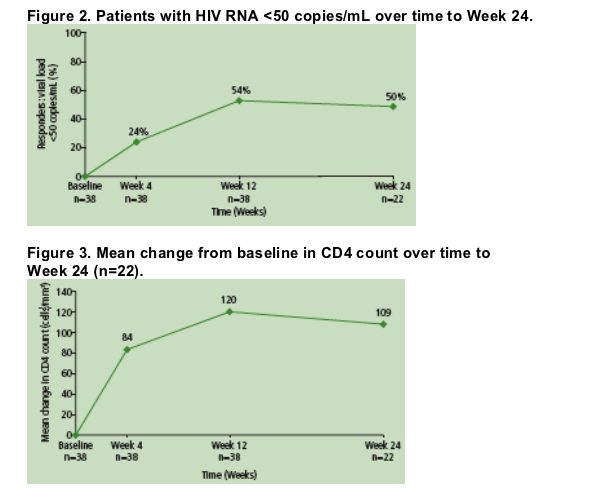
54% of patients achieved HIV RNA <50 copies/mL at Week 12 (n=38), and 50% had HIV RNA <50 copies/mL (n=22) atWeek 24 (Figure 2).
AtWeek 24, mean increase from baseline in CD4 count was 109 cells/mm3 (n=22) (Figure 3).
Among 22 patients with CD4 data at both baseline and Week 24, 10 had a CD4 count <50 cells/mm3 at baseline. By Week 24, 80% of these 10 patients had a CD4 count >100 cells/mm3 (Figure 4).
Safety
DRV/r was generally safe and well tolerated. Most AEs were mild to moderate in severity.
9 AEs were considered possibly related to DRV:
- 2 mild rashes and 1 mild case of nausea.
- 2 increased ALT (Grade 2), 1 hypercholesterolemia (Grade 2),
1 hypertriglyceridemia (Grade 3), 1 increased creatinine (Grade 2), and 1
increased total bilirubin (Grade 2).
Two serious AEs (ENF injection-related hematoma requiring surgery and 1 death) were considered unrelated to DRV/r.
Conclusions
In our single-center experience, DRV/r 600/100mg bid provided clinically meaningful decreases in VL and increases in CD4 count through Week 24.
Most patients who achieved undetectable VL (<50 copies/mL) did so within the first 12 weeks of treatment, and responses were maintained through 24 weeks.
DRV/r was well tolerated, with infrequent AEs; no unexpected events were reported.
Reference
1. De Meyer S, et al. Antimicrob Agents Chemother. 2005;49:2314-2321.
|
|
| |
| |
|
|
|