 |
|
|
| |
Good News and Mixed News on New Antiretrovirals: Integrase and CCR5 Inhibitors, a Nuke, and a Nonnuke
|
| |
| |
46th ICAAC, September 27-30, 2006, San Francisco
Mark Mascolini
Without question the top HIV news from the 46th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) involved pipeline antiretrovirals. While inhibitors of HIV integrase and the CCR5 coreceptor on T cells won most headlines, this yearly get-together also featured important findings on a nucleoside (NRTI) with an extra-long half-life and the resistance-busting nonnucleoside (NNRTI) etravirine (TMC125).
Integrase inhibitor stalls multiresistant HIV
Everyone sounds excited about MK-0518, the investigational Merck integrase inhibitor, and two ICAAC reports only fanned the flames of this ardor. In people with profound antiretroviral experience and few--or no--antiretrovirals that promised to buck up a salvage regimen, MK-0518 stalled HIV impressively [1]. It worked even better when enfuvirtide-naive people could start that fusion inhibitor with the Merck drug. The second study showed that 24 weeks of MK-0518 (plus 3TC and tenofovir) has little effect on lipids, except that triglycerides fell in people taking the highest dose of the integrase inhibitor [2].
Beatriz Grinsztejn (Evandro Chagas Clinical Research Institute, Rio de Janeiro) and colleagues in Europe and the US randomized 178 treatment-experienced people to 200, 400, or 600 mg of MK-0518 twice daily, or placebo, plus an optimized background regimen picked on the basis of resistance tests and treatment history [1]. But the researchers could do little or nothing to optimize the new regimens of most study participants. About half of those enrolled had a phenotypic sensitivity score (PSS) of 0 to all antiretrovirals, meaning the Phenosense GT assay rated their virus resistant to every available drug except MK-0518 (Table 1). According to this same test, about 90% of study participants did not have a single active protease inhibitor (PI) to put in their new regimen.
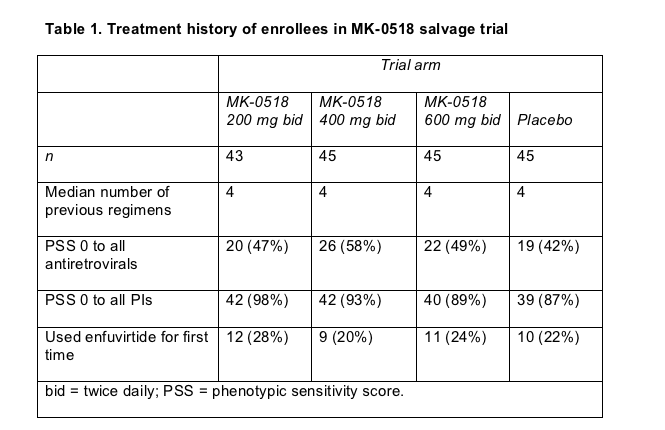
When people signed up for the trial their viral loads averaged 4.6 to 4.8 log copies/mL (about 40,000 to 63,000 copies) across the study arms. Average CD4 counts stood at 200 to 245 in the three MK-0518 arms and at 274 in the placebo group.
By study week 24, 10 people (23%) had quit the 200-mg arm, 8 people (18%) had left the 400-mg arm, 8 people (18%) the 600-mg arm (18%), and 31 people (69%) the placebo arm. Three dropouts in the MK-0518 arms and 1 in the placebo arm resulted from clinical problems or side effects. Lack of efficacy caused the other 49 dropouts.
A 24-week noncompleter-equals-failure analysis figured that about 70% in all three MK-0518 groups had a viral load below 400 copies, compared with fewer than 20% in the placebo group. While 57% to 67% in the MK-0518 arms had a 24-week load below 50 copies, only 14% in the control arm hit that mark. About 80% taking MK-0518 at any dose had at least a 1-log (10-fold) viral load drop by week 24, compared with 18% in the placebo arm. Viral load declines averaged about 2 log (100-fold) with the integrase inhibitor and 0.4 log with placebo plus other background drugs. Among people who started the trial with a phenotypic sensitivity score of 0, 54% to 62% randomized to MK-0518 had a 24-week viral load under 400 copies/mL, while no one in the placebo group with a baseline score of 0 got below 400 copies (Table 2).
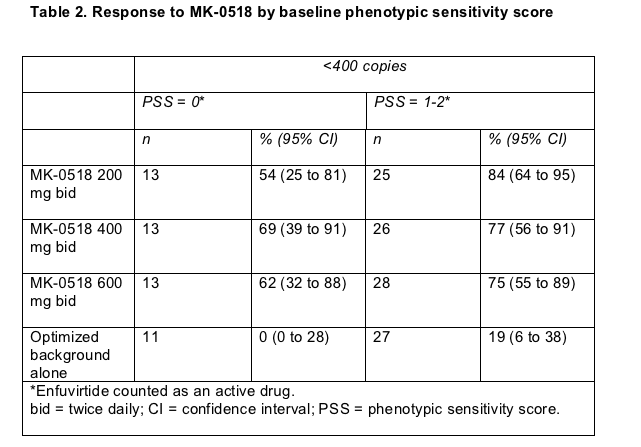
As in previous salvage trials, putting the fusion inhibitor enfuvirtide in the regimen boosted chances of virologic response. Among people in the placebo arm, the percentage with a 24-week load below 400 copies rose from about 15% without enfuvirtide to about 30% with the injectable drug. In the MK-0518 arms the response rate climbed from about 60% without enfuvirtide to 90% or more with enfuvirtide. These findings underline the advantage of waiting until one has at least two potent antiretrovirals for a salvage regimen--if at all possible--rather than adding new drugs one at a time to already enfeebled antiretrovirals.
Grade 3 or 4 lab abnormalities proved uncommon and their rates were similar in the MK-0518 and placebo groups. Grinsztejn reported four serious clinical setbacks--one stroke in the placebo group, one case of acute pancreatitis after two doses in the 200-mg MK-0518 group (attributed to the background regimen), one case of lipoatrophy, and one death from metabolic acidosis and renal insufficiency.
What MK-0518 does (or doesn't) do to lipids
This new integrase inhibitor also looked good in a trial enrolling previously untreated people [3]. At the 24-week point MK-0518 plus 3TC and tenofovir controlled HIV as well as efavirenz plus 3TC and tenofovir and appeared to do so faster. At ICCAC, Merck unveiled lipid trends from this trial showing that MK-0518 had little or no ill effects on circulating fats through 24 weeks.
The lipid analysis involved 184 people with no antiretroviral experience randomized to take 100, 200, 400, or 600 mg of MK-0518 twice daily or efavirenz plus the two NRTIs. Pretreatment cholesterol and triglycerides were equivalent across the five study arms and in the normal range (Table 3) in this relatively young study group (median age 34 to 37 years across the five arms).
By study week 24 average total cholesterol and triglycerides drifted slightly downward in all MK-0518 groups while rising among people taking efavirenz (Table 3). Among people assigned to the highest MK-0518 dose, 600 mg twice daily, triglycerides sunk by an average 43 mg/dL after 24 weeks of treatment, compared with a 47-mg/dL hike in the efavirenz group.
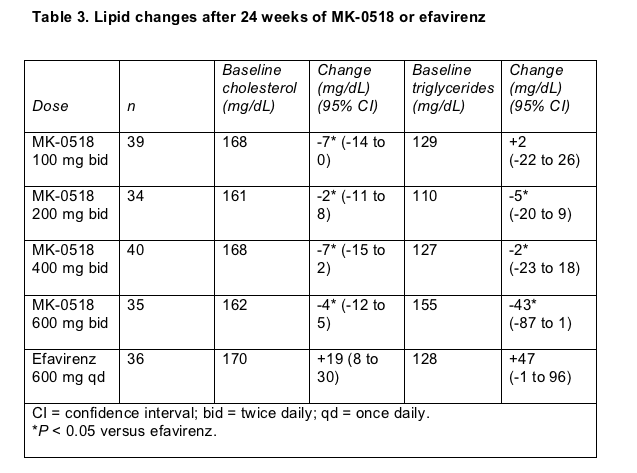
Most of the cholesterol gained with efavirenz was "good" high-density lipoprotein (HDL) cholesterol. Efavirenz takers added an average of about 4 mg/dL of HDL cholesterol in 24 weeks, while most of the MK-0518 groups gained about 2 mg/dL; these differences lacked statistical significance.
"Bad" low-density lipoprotein (LDL) cholesterol fell slightly in all four MK-0518 arms while edging marginally upward with efavirenz. All these LDL differences between the MK-0518 groups and the efavirenz group were statistically significant (P < 0.05) but probably not clinically meaningful at this point of the trial because the efavirenz group gained an average of only about 5 mg/dL from a starting level of 108 mg/dL.
Other ICAAC studies probed for potential MK-0518 interactions with other antiretrovirals. Multiple doses of tipranavir/ritonavir slightly trimmed the 12-hour concentration of MK-0518 given to healthy volunteers at 400 mg twice daily but did not greatly affect the area under the concentration-time curve (AUC) or maximum concentration of MK-0518 [4]. The same dose of MK-0518 had no significant interactions with tenofovir [5]. A third study in healthy volunteers found that 400 mg of MK-0518 modestly reduces the AUC, 12-hour concentration, and maximum concentration of efavirenz [6]. But lack of any effect on the time to maximum concentration of efavirenz or efavirenz half-life led Merck investigators to conclude that MK-0518 will not have a "clinically meaningful" interaction with efavirenz. In this study MK-0518 did not affect levels of ritonavir given at 100 mg.
Is there anything bad to report about MK-0518? Not yet. At an International Association of Physicians in AIDS Care meeting just before ICAAC, Brian Gazzard, the research chief at London's Chelsea and Westminster Hospital, cited speculation that the integrase inhibitor may have unforeseen long-term side effects because--unlike other antiretrovirals--it "fiddles with" the CD4-cell nucleus. But so far he finds response data with MK-0518 "extremely impressive." Gazzard mulled a new sequencing paradigm in which treatment would begin with fixed-dose tenofovir/emtricitabine or 3TC/abacavir plus a nonnucleoside, followed by two different nucleosides plus either darunavir/ritonavir or an integrase inhibitor.
Speaking at the same meeting, Anthony Fauci (National Institute of Allergy and Infectious Diseases, Bethesda) had some happier speculation to air. Because HIV becomes latent (and thus impervious to antiretrovirals) when it integrates with CD4-cell DNA, integrase inhibitors may help prevent the buildup of latent reservoirs by barring integration. Whether limiting latent pool size has any clinical benefit is another question. But if one buys Fauci's hypothesis and believes in keeping latent pools low, it would be hard to resist putting an integrase inhibitor into a person's first regimen.
Integrase inhibitor resistance and cross-resistance
MK-0518 is not the only integrase inhibitor now running the clinical trial gauntlet. GS-9137, a Gilead Sciences candidate in phase 2 studies, has at least three unique features in the still-young integrase inhibitor class: It appears to work well in a once-daily dose; it benefits from a ritonavir boost; and it may be the only antiretroviral candidate backed by a cigarette company (Japan Tobacco). In a placebo-controlled trial involving 40 antiretroviral-naive or -experienced people, GS-9137/ritonavir at a dose of 50/100 mg once daily pushed viral loads down 100-fold--without help from other antiretrovirals [7].
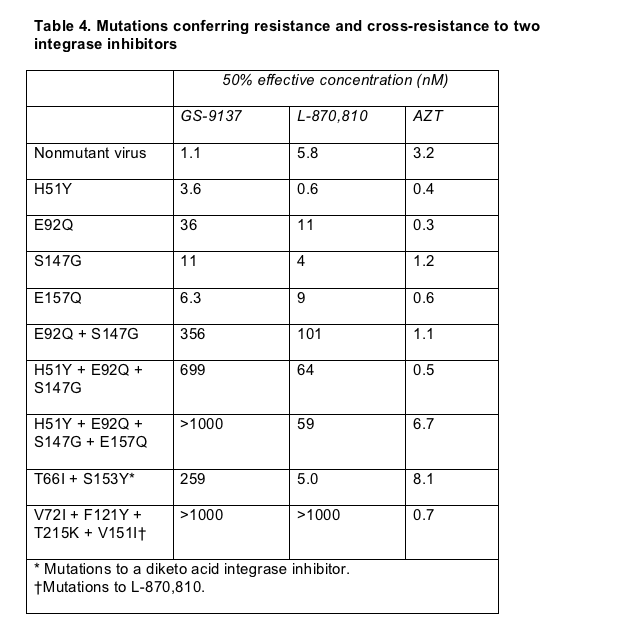
At ICAAC researchers from Japan Tobacco and Kyoto's Institute for Virus Research offered a look at what may happen when HIV becomes resistant to an integrase inhibitor [8]. Standard serial passage studies, which douse HIV-infected cultures with slowing rising doses of an antiretroviral, found that a single mutation can render HIV resistant to GS-9137 and that virus resistant to this integrase inhibitor may be resistant to others.
After 30 passages (or exposures to GS-9137), HIV evolved an E92Q mutation in the integrase catalytic core domain. After 60 passages H51Y and S147G joined E92Q, and at passage 70 E157Q arose. Next the researchers tested susceptibility of virus bearing these mutations to GS-9137, an earlier Merck anti-integrase drug labeled L-870,810 [9], and AZT. By itself the E92Q substitution made HIV resistant to the Gilead integrase inhibitor and cross-resistant to L-870,810 (Table 4). Virus engineered to carry two or three of the identified mutations proved highly resistant to GS-9137 and L-870,810 in this assay. Medleys of different mutations discovered earlier in studies of two Merck integrase inhibitor candidates also made HIV highly resistant to GS-9137.
In separate studies the Japan Tobacco and Kyoto team showed that virus isolated from 4 people with antiretroviral experience remained highly susceptible to GS-9137.
No one can predict the clinical consequences of these cross-resistance findings; what happens in serial passage studies doesn't necessarily happen in people. But if a single mutation makes HIV resistant to one integrase inhibitor and cross-resistant to others, they would have to be prescribed with the same caution taken in prescribing nonnucleosides. And if HIV resistant to one integrase inhibitor stands a high chance of resistance to others, physicians will want to prescribe the best (most potent, least toxic, most convenient) integrase inhibitor first.
Plugging CCR5 with a monoclonal antibody
Most attention on blocking CCR5 coreceptors has focused on the CCR5 antagonists--aplaviroc (no longer being developed), maraviroc, and vicriviroc. And ICAAC featured some intriguing studies on all three of those drugs. But a humanized IgG4 monoclonal antibody under study at Human Genome Sciences also keeps HIV off CCR5 receptors and boosts the power of traditional antiretrovirals [10]. Twenty-four weeks of serial passage pressure did not promote emergence of mutations conferring resistance to this agent, but that doesn't mean HIV can't find a way to outmaneuver it.
A disadvantage of this monoclonal antibody, still enigmatically tagged CCR5mAb004, is that it requires intravenous infusion. In a study presented at ICAAC by Jay Lelezari (Quest Clinical Research, San Francisco), 54 antiretroviral-naive people with virus that homes exclusively to CCR5 and with more than 5000 HIV RNA copies and more than 250 CD4s got randomized to a single infusion of 0.4, 2, 8, 20, or 40 mg/kg of 004 or placebo [11]. The 2-mg/kg dose raised a moderately severe urticarial rash in 2 people. After that the researchers pretreated people with oral diphenhydramine (Benadryl) and no more rashes flared. No one had grade 3 or 4 treatment-related problems.
This agent has a slow onset of action--3 to 4 days--while it coats CCR5 receptors. But one infusion lasts a long time. The monoclonal antibody eventually saturated more than 80% of receptors 14 to 28 days after infusion of 8 mg/kg or higher doses. Lalezari calculated a 5- to 8-day half-life for 004. Maximum concentration of the monoclonal antibody was dose-proportional (the higher the dose, the higher the concentration), and area under the concentration-time curve (AUC) was more than dose-proportional.
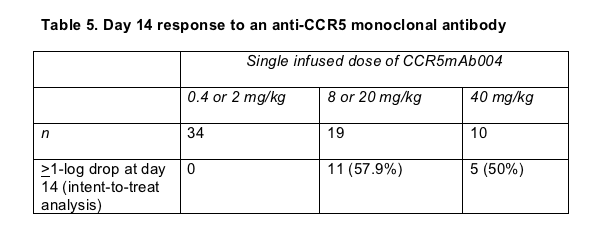
More than half of the people who took the higher doses of 004 had at least a 1-log (10-fold) drop in viral load at day 14 (Table 5). People who began therapy with a lower pretreatment viral load had a better chance of knocking their load down farther: Pretreatment loads averaged 4.0 log (10,000 copies) in people with more than a 1-log drop and 4.4 log (about 25,000 copies) in people with less than a 1-log drop (P = 0.02). The monoclonal antibody's 90% inhibitory concentration was significantly lower (better) in people who had at least a 1-log viral load dip (31.3 ug/mL) than in those who did not (87.5 ug/mL) (P = 0.0388).
Notably, HIV preference for CCR5 changed in 5 people after 1 day of treatment. Because the Monogram Biosciences assay used to determine coreceptor preference (tropism) still has shortcomings, it's impossible to say whether these coreceptor shifts resulted from infusion of the monoclonal antibody or whether they simply reflect the assay's inadequacies. Whatever the explanation, none of the 5 people with changing tropism had a good virologic response to 004, even though 4 of them got either the 20- or 40-mg/kg dose.
How this infused monoclonal antibody would be integrated into an oral antiretroviral regimen and whether it can compete with oral CCR5 antagonists remain to be determined.
Similar response with R5-only and R5/X4 mixed virus
CCR5-tropic HIV dominates a person's viral swarm in the early months to years of HIV infection, though work reported last year showed that viral populations using either CCR5 or CXCR4 (dual/mixed-tropic virus) may appear at any CD4 count or viral load [12]. This study by Graeme Moyle and Chelsea and Westminster Hospital colleagues found, however, that strictly CCR5-tropic virus becomes less common as CD4 counts wane and viral loads rise.
At ICAAC, Laura Waters in Moyle's group reported that CD4 counts fall faster--for awhile--in antiretroviral-naive people with dual/mixed-tropic HIV than in those with strictly CCR5-favoring virus [13]. This analysis involved 402 untreated people, 326 with R5-tropic virus, 73 with dual/mixed-tropic HIV, and 3 with virus that uses only CXCR4, according to the Monogram assay. Checking stored samples from these people, Waters found a significantly higher median CD4 count in the first samples of the R5-only group than in the first samples of the dual/mixed group (325 versus 203, P < 0.001). Likewise, baseline median viral loads were significantly lower in the strictly R5-tropic group than in those with dual- or mixed-tropic HIV (39,385 versus 142,568 copies, P < 0.001).
From this baseline measure to 12 months before these people started a potent antiretroviral regimen (which excluded triple-NRTI regimens), CD4s fell significantly faster in those with dual/mixed-tropic virus than in those with CCR5-only virus (P < 0.026) in an analysis adjusted for first viral load. But in the 2 years before treatment began, CD4 declines did not differ significantly by coreceptor tropism (she said there were dropouts in the X4 group).
Once treatment did start, coreceptor preference had no impact on response to antiretrovirals among people who had at least 6 months of therapy. CD4 gains averaged 185 in the R5-only group and 182 in the dual/mixed group 12 months after antiretrovirals began. Average gains after 24 months measured 247 in the R5-only group and 292 in the dual/mixed group. Virologic response also proved similar in the two groups after 6, 12, or 24 months of therapy.
These results contradict findings of a briefer Swiss HIV Cohort Study analysis that recorded a significantly smaller CD4 gain in the first 6 months of therapy among ART-naïve people with R4- or mixed-tropic virus [14]. CD4 counts rose by an average 82 cells in people with R5-only virus at baseline and by 40 cells in those with mixed-tropic virus at baseline (P = 0.012). Respective 6-month CD4 climbs in people with R5-only versus mixed tropism at follow-up measured 90 and 35 cells (P = 0.049). People with mixed tropism at baseline had a 4 times higher risk of clinical progression during treatment, and people with mixed tropism during follow-up had a 10.3 times higher risk in an analysis adjusted for other variables that influence progression.
What explains the difference between the findings of these two studies? Three possibilities are the longer on-treatment follow-up in the Chelsea and Westminster study, different approaches to these analyses (the Swiss compared 96 progressors with 84 nonprogressors), and a different assay to gauge coreceptor tropism (the Swiss used their own assay rather than the Monogram test).
At this point it's hard to say which team's results point in the right direction. The Chelsea and Westminster findings make sense in that current regimens can control replication of both CCR5- and dual/mixed-tropic virus. The Swiss results make sense in that dual/mixed-tropic virus shows up in more advanced HIV infection and people with more advanced infection may have a slower or worse response to first therapy.
Tropism may differ in blood and brain
Tests to tell whether a person's viral population prefers CCR5 or CXCR4--or whether it can use either coreceptor--typically scrutinize virus reaped from blood samples. But do viral coreceptor leanings differ in HIV collected from blood versus cerebrospinal fluid (CSF) of the same person?
To find out, researchers from Lund University and Sahlgrenska Institute in Sweden compared tropism in paired plasma and CSF samples from 28 people at different stages of HIV infection [15]. Part of this analysis relied on lab-constructed chimeric receptors these researchers engineered to include parts of both CCR5 and CXCR4 [16]. They used virus stocks to infect cells brandishing CD4 and either CCR5, CXCR4, or these R5/X4 chimeras.
R5-favoring virus proved dominant in both plasma and CSF from these 28 people--with some exceptions. Plasma virus from 7 people used either X4 alone (in 1 person) or X4 and R5 (in 6 people). But CSF from 4 of these 7 people harbored only R5-tropic HIV. Among 21 people whose plasma and CSF virus homed exclusively to CCR5, 6 had major differences in chimeric coreceptor use. In total, 10 of 28 people (36%) had evidence of discordant coreceptor tropism in plasma and CSF.
The ability of plasma R5 virus to use the R5/X4 chimeric receptor labeled FC-2 strongly correlated with greater CD4 deficits but not with higher viral loads. Nine people with FC-2 use and exclusive R5 isolates in plasma had a median CD4 count of 49--versus 495 CD4s in 12 people with FC-2-negative virus (P = 0.004). As earlier studies show, X4 virus or mixed R5/X4 virus signaled advanced CD4 depletion (median 60 cells, P = 0.005 versus the FC-2-negative group).
These researchers suggest that evolution of different coreceptor tropism in blood and CSF "most likely reflects more efficient [viral] replication in abundant target [CSF] cells," such as macrophages and microglial cells. They propose that "discordance of viral phenotype in plasma and CSF is frequent and needs to be considered in the context of emergent treatment" with CCR5 antagonists.
A literature search turned up a solitary study looking at how a CCR5 antagonist affects HIV load in CSF [17]. Although the Swedish study found CCR5-tropic virus dominating most patients' CSF population [15], this University of Miami trial determined that a CCR5 antagonist candidate called D-Ala1-peptide T-amide (DAPTA) harnessed HIV in CSF no better than placebo. That finding strongly suggests DAPTA does not penetrate the blood-brain barrier, because it did lower viral loads in blood. Whether the same proves true of maraviroc and vicriviroc remains unknown--or unreported.
Viral susceptibility and coreceptor occupancy
GlaxoSmithKline suspended development of its CCR5 antagonist, aplaviroc, when a few people taking it suffered severe liver toxicity. But an aplaviroc study reported at ICAAC may hold clues to the efficacy of Pfizer's maraviroc and Schering's vicriviroc.
Glaxo's James Demarest offered data indicating that resistance to aplaviroc in people with virologic failure could prove hard to spot with standard viral-population-based testing [18]. He also used clonal analysis--which looks at a wide array of clones created from HIV isolates--in 10 antiretroviral-naive people who began treatment with aplaviroc plus lopinavir/ritonavir and endured virologic failure, defined as less than a 1-log (10-fold) drop in viral load by treatment week 4, a confirmed load above 400 copies after a sub-400 load, or more than a 0.5-log viral load climb from the lowest recorded load.
Testing on the population level detected no drop in susceptibility to either aplaviroc or lopinavir. But clonal analysis uncovered reduced susceptibility to aplaviroc in fewer than half of the clones derived from 6 of the 10 people. Demarest also found shifts in coreceptor affinities from R5-only virus to R5/X4-mixed virus in 2 of these 10. But phylogenetic analysis suggested that 1 person had mixed-tropic virus before treatment began. The study did not turn up mutations in the viral envelope gene that correlate with reduced susceptibility to aplaviroc.
Schering reported cell study results gauging how avidly four CCR5 antagonists bind to their target receptor, and how long they stick to the target after binding [19]. Maraviroc and aplaviroc bound CCR5 more tightly than vicriviroc of SCH-C, an earlier Schering candidate (Table 6). But vicriviroc clung to CCR5 almost twice as long as maraviroc (Table 6). Aplaviroc won the durability contest, plugging CCR5 receptors twice as long as vicriviroc.
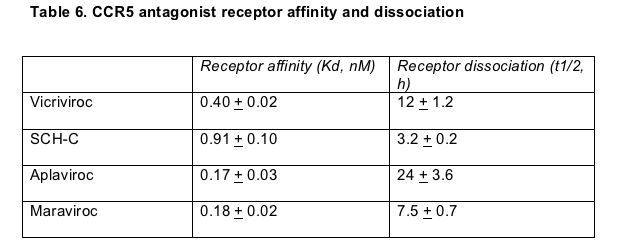
Vicriviroc's slow receptor dissociation rate supports its once-daily dosing schedule; ongoing maraviroc trials are testing both once- and twice-daily dosing. Whether this dissociation advantage and potential dosing plus for vicriviroc translate into a clinical edge can be determined only in a head-to-head comparison with maraviroc.
Elvucitabine monotherapy: new nuke with staying power
The nucleoside field has grown thick with once-daily options, and one more, elvucitabine, may be on the way. In fact plasma and peripheral blood mononuclear cell (PBMC) half-lives of this NRTI are so lengthy it may be a candidate for less-than-daily dosing.
Previous work showed that 21 days of elvucitabine at doses of 5 or 10 mg daily or 20 mg every other day, plus lopinavir/ritonavir, trimmed viral loads 1.8, 1.9, and 2.0 log copies/mL in antiretroviral-naive people [20]. (Earlier studies of a 50-mg dose found evidence of bone toxicity.) To gauge how well elvucitabine slows HIV replication on its own, Philippe Colucci (University of Montreal) and colleagues at Achillion Pharmaceuticals gave 10 mg of the drug or placebo once daily to 24 treatment-naive people for 7 days [21]. From days 8 through 21, people taking elvucitabine stopped the drug and started lopinavir/ritonavir to forestall emergence of resistance to the long-lasting NRTI. Virus resistant to elvucitabine or lopinavir/ritonavir did not emerge in the trial.
Pretreatment viral loads averaged 4.76 log (about 57,500 copies) in the elvucitabine arm and 4.72 log (about 52,500 copies) in the placebo group. Baseline CD4 counts averaged 320 (range 201 to 478) in the elvucitabine group and 403 (range 111 to 641) in the placebo arm. After 7 days of elvucitabine monotherapy, the average viral load dropped 0.85 log, while CD4 counts rose by an average 62 cells. Both improvements were statistically superior to changes in the placebo group.
Although people stopped elvucitabine after day 7, levels of the nucleoside stayed above the 50% inhibitory concentration through day 21. At that point the maximum viral load drop measured 1.72 log. Colucci estimated an elvucitabine half-life of about 100 hours in both plasma and PBMCs. The only side effects were mild diarrhea and rising triglycerides, both noted after people started lopinavir/ritonavir.
One-two punch from the two TMCs
Two single-center studies suggest that the PI darunavir (TMC114) and the investigational NNRTI etravirine (TMC125) could be a formidable duo for people with virus resistant to other drugs in those classes. Thirteen of 15 people in these small, nonrandomized studies--one presented at ICAAC and the other at the August International AIDS Conference--got their viral loads under 50 copies within 24 weeks of starting darunavir and etravirine with a clutch of nucleosides and, sometimes, enfuvirtide.
At London's Chelsea and Westminster Hospital, Marta Boffito and coworkers paired the TMC drugs in 12 people with woeful resistance records [22]. Two people withdrew consent for personal reasons. Among the remaining 10, the median number of primary PI mutations stood at 4 (range 0 to 5), the median number of NNRTI mutations at 2 (range 0 to 6), and the median number of NRTI mutations at 7 (range 2 to 9). Median fold-change in susceptibility to PIs ranged from 0.4 to 137.6 (1.25 for darunavir) and to NNRTIs from 0.6 to 104.9 (1.25 for etravirine). Six of 10 people had already tried tipranavir and emtricitabine; only 2 people started emtricitabine as a new drug in this study. Everyone took 200 mg of etravirine twice daily, 600/100 mg of darunavir/ritonavir twice daily, and two or more nucleosides.
After 24 weeks 9 of 10 people had a viral load under the 50-copy mark. The tenth had a load of 722 copies, which dwindled to 59 copies by week 32. Median drop in HIV RNA log copies/mL measured 2.7 (range 2.3 to 3.9), and median CD4 gain stood at 113 (range 41 to 268) cells. No one had a serious clinical setback or dangerous lab reading. Possible treatment-related side effects included mild diarrhea in 1 person, headache in 1, and rash in 1. All problems resolved without a change in drugs or dosing.
In Vancouver, Julio Montaner and colleagues tried the same regimen in 5 heavily experienced people [23]. All had primary PI and NNRTI mutations, and 3 had tried enfuvirtide. After 24 weeks of double-TMC salvage, 4 of 5 people registered a sub-50-copy viral load. The one person who did not reach an undetectable load was one of the three who had already used enfuvirtide.
A study of 8 HIV-infected people taking two nucleosides plus
fosamprenavir/ritonavir at a dose of 700/100 mg twice daily found that the PIs did not affect levels of etravirine after they added the nonnucleoside to their regimen at a dose of 800 mg twice daily [24]. (The newer etravirine formulation achieves the same drug exposure with 200 mg twice daily.) But etravirine boosted amprenavir's AUC 69%, maximum concentration 62%, and minimum concentration 77%. (Amprenavir is the metabolite of fosamprenavir.) Ritonavir levels did not change with etravirine. No one had serious or grade 3 or 4 side effects. Tibotec researchers suggested that fosamprenavir's dose may have to be lowered when boosted by ritonavir and given with etravirine. A separate study in healthy volunteers found no need for dose adjustments when combining etravirine with tenofovir [25].
Mark Mascolini writes about HIV infection (markmascolini@earthlink.net).
References
1. Grinsztejn B, Nguyen B, Katlama C, et al. Potent efficacy of MK-0518, a novel HIV-1 integrase inhibitor, in patients with triple-class resistant virus: 24-week data. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1383.
2. Teppler H, Azrolan N, Chen J, et al. Differential effect of MK-0518 and efavirenz on serum lipids and lipoproteins in antiretroviral therapy (ART)-naive patients. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-256a.
3. Markowitz M, Nguyen BY, Gotuzzo F, et al. Potent antiretroviral effect of MK-0518, a novel HIV-1 integrase inhibitor, as part of combination ART in treatment-naive HIV-1 infected patients. XVI International AIDS Conference. August 13-18, 2006. Toronto. Abstract THLB0214.
4. Wenning LA, Hanley H, Stone J, et al. Effect of tipranavir + ritonavir on pharmacokinetics of MK-0518. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract A-374.
5. Wenning LA, Friedman E, Kost JT, et al. Lack of a significant drug interaction between MK-0518 and tenofovir disoproxil fumarate. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract A-375.
6. Iwamoto M, Wenning LA, Petry AS, et al. Minimal effect of ritonavir and efavirenz on pharmacokinetics of MK-0518. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract A-373.
7. DeJesus E, Berger D, Markowitz M, et al. The HIV integrase inhibitor GS-9137 (JTK-303) exhibits potent antiviral activity in treatment-naive and experienced patients. 13th Conference on Retroviruses and Opportunistic Infections. February 5-8, 2006. Denver. Abstract 160LB.
8. Kodama E, Shimura K, Sakagami Y, et al. In vitro antiviral activity and resistance profile of a novel HIV integrase inhibitor JTK-303/GS-9137. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-254.
9. Little S, Drusano G, Schooley R, et al. Antiretroviral effect of L-000870810, a novel HIV-1 integrase inhibitor, in HIV-1-infected patients. 12th Conference on Retroviruses and Opportunistic Infections. February 22-25, 2005. Boston. Abstract 161.
10. Giguel F, Beebe L, Migone TS, Kuritzkes D. The anti-CCR5 mAb004 inhibits HIV-1 replication synergistically in combination with other antiretroviral agents but does not select for resistance during in vitro passage. 13th Conference on Retroviruses and Opportunistic Infections. February 5-8, 2006. Denver. Abstract 505.
11. Lalezari J, Lederman M, Yadavalli G, et al. A phase 1, dose-escalation, placebo-controlled study of a fully human monoclonal antibody (CCR5mAb004) against CCR5 in patients with CCR5 tropic HIV-1 infection. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1668.
12. Moyle GJ, Wildfire A, Mandalia S, et al. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J Infect Dis 2005;191:866-872.
13. Waters LJ, Mandalia S, Wildfire A, et al. CXCR4/mixed-tropic HIV-1 is associated with more rapid CD4 cell decline compared with CCR5-tropic virus in antiretroviral naive individuals. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1667.
14. Philpott S, Ledergerber B, Klimkait T, et al. CXCR4-specific viral load predicts clinical HIV-1 disease progression during HAART. 3rd IAS Conference on HIV Pathogenesis and Treatment. July 24-27, 2005. Rio de Janeiro. Abstract THAA0201.
15. Karlsson UJ, Antonsson L, Owman C, et al. Coreceptor use in paired plasma and cerebrospinal fluid HIV-1 isolates. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-246.
16. Karlsson I, Antonsson L, Shi Y, et al. HIV biological variability unveiled: frequent isolations and chimeric receptors reveal unprecedented variation of coreceptor use. AIDS 2003;17:2561-2569.
17. Goodkin K, Vitiello B, Lyman WD, et al. Cerebrospinal and peripheral human immunodeficiency virus type 1 load in a multisite, randomized, double-blind, placebo-controlled trial of D-Ala1-peptide T-amide for HIV-1-associated cognitive-motor impairment. J Neurovirol 2006;12:178-189.
18. Madsen H, Kitrinos K, Irlbeck D, et al. Detection of reduced susceptibility to aplaviroc when administered with lopinavir + ritonavir requires clonal analysis of HIV-1 envelope. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1669.
19. Gonsiorek W, Strizki JM, Hesk D, et al. Analysis of binding kinetics and coupling states reveals distinct binding properties of small molecule CCR5 antagonists. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-243.
20. Colucci P, Pottage J, Robison H, et al. The different clinical pharmacology of elvucitabine (beta-L-Fd4C) enables the drug to be given in a safe and effective manner with innovative drug dosing. 45th ICAAC. December 16-19, 2005. Washington, DC. Abstract LB-27.
21. Colucci P, Pottage J, Robison H, et al. Efficacy and novel pharmacology of elvucitabine in a 7-day placebo-controlled monotherapy study. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1670d.
22. Boffito M, Winston A, Jackson A, et al. Week 24 results and baseline phenotypic susceptibility in treatment-experienced patients initiating the combination of TMC114/r and TMC125. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1000.
23. Montaner J, Harris M, Kakuda G, et al. Combination of TMC114/r and TMC125 in patients with multidrug-resistant HIV. XVI International AIDS Conference. August 13-18, 2006. Toronto. Abstract THPE0136.
24. Scholler-Gyure M, Woodfall B, Bollen S, et al. Pharmacokinetics of amprenavir and TMC125 in HIV-infected volunteers receiving TMC125 with fosamprenavir/ritonavir. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract A-370.
25. Scholler-Gyure M, Debroye C, Woodfall B, et al. Pharmacokinetic evaluation of the interaction between TMC125 and tenofovir disoproxil fumarate. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract A-371.
|
| |
|
|
|
|
|