 |
|
|
| |
Resistance Before Therapy, Hidden Mutations, and Blips
|
| |
| |
46th ICAAC, September 27-30, 2006, San Francisco
Mark Mascolini
ICAAC featured a diverse array of reports on resistance to antiretrovirals, many of them from industry researchers sifting returns from big clinical trials or Monogram's vast resistance database. Attendees learned that resistance rates are still on the rise among untreated Americans, how occult mutations affect response to Trizivir plus tenofovir, and whether shaky adherence explains viral load blips.
US resistance rates still rising in untreated
Infection with resistant HIV, especially virus resistant to nonnucleosides (NNRTIs), continues to rise across the United States, according to results of a 2035-person study by Lisa Ross of GlaxoSmithKline [1]. Because of climbing NNRTI resistance in untreated people, Ross recommended first-line protease inhibitor (PI) therapy in regions with particularly high NNRTI resistance rates--unless a person's virus can be tested for resistance mutations before treatment.
The Glaxo study involved 2035 antiretroviral-naive people who enrolled in clinical trials from 2000 to 2005 in 33 states and Washington, DC. Most cohort members, 84%, were men, and more than 90% became infected during sex. Median viral load and CD4 count when people enrolled in the trials were 4.86 log copies/mL and 252 cells.
Figuring mutation rates according to the IAS-USA list of primary mutations and the Stanford database, Ross found little difference between the systems. From 2000 through 2005, prevalence of infection with resistant virus stood at 11% according to both the IAS-USA and Stanford schemes. NNRTI mutations proved most common by the IAS-USA system (6%), followed by nucleoside (NRTI) mutations (4%), and PI mutations (3%).
Prevalence of any pretreatment mutation rose from about 5% in 2000, to 10% in 2001, to a peak above 15% in 2005. From 2000 to 2005 the risk of any IAS-USA mutation climbed more than 20% (odds ratio [OR] 1.209, P < 0.0001) and the risk of any NNRTI mutation surged 21% (OR 1.213, P = 0.0011).
Over the 5 study years the risk of any IAS-USA resistance mutation jumped 12% for whites, 12% for Hispanics, and 8% for blacks. Multivariate analysis determined that, compared with whites, blacks had a significantly lower chance of infection with resistant virus from any class according to either IAS-USA (P = 0.0033) or Stanford definitions (P = 0.0012). The same held true for each drug class figured separately. The Glaxo team did not speculate on reasons for this discrepancy. Perhaps the most likely explanation is that fewer African Americans enrolling in these trials got infected by people taking antiretrovirals.
Among people with a pretreatment viral load below 100,000 copies, multivariate analysis figured an ever-escalating risk of infection with resistant virus from 2000 to 2005. Over that period people ran a 26% greater risk of becoming infected with a higher number of IAS-USA mutations (OR 1.259, P = 0.0009) and a 19% greater risk of infection with more Stanford-defined mutations (OR 1.192, P = 0.0089). These significantly higher risks in later years held true in separate analyses of NNRTI mutations and NRTI mutations, but not PI mutations.
Ross and colleagues concluded that the high primary resistance rates they recorded support US Department of Health and Human Services recommendations to genotype virus before treating people with acute or chronic HIV infection. The heightened risk of infection with NNRTI-resistant virus, confirmed in other recent studies,[2-4] led Ross to suggest that "if resistance testing is not an option, then use of an NNRTI-sparing first-line regimen should be considered, especially in areas such as New York, where a 13.4% incidence of transmitted NNRTI resistance has been reported for 2003-2004 [5]."
In a separate analysis of the Monogram resistance database, Ross found that 33.6% of viral isolates with no primary NRTI mutations had decreased susceptibility to one or more NNRTIs [6]. Because the 26,852 isolates collected from 2003 through 2005 were not linked to patient records, Ross could not tell how many of the NNRTI-resistant viruses came from treated versus untreated people. But recent work showing NNRTI resistance prevalence of 7% to 13% in untreated people [2-5] suggests that the larger portion of NNRTI-resistant isolates in the current study came from treated people.
Among isolates with one primary NRTI mutation, 53.8% had reduced susceptibility to an NNRTI, and among those with two primary NRTI mutations, 66.6% had resistance to an NNRTI. Rates of resistance to PIs were much lower in isolates with 0, 1, or 2 primary NRTI mutations (Table 1).
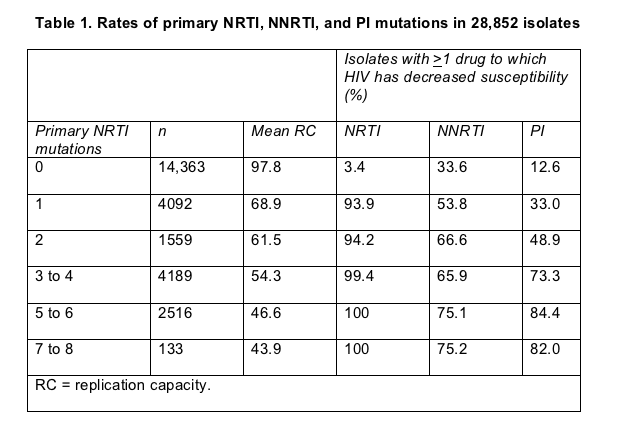
This study also traced a plummet in viral replication capacity after a single NRTI mutation evolves (Table 1), perhaps, Ross speculated, because M184V/I and K65R evolve early in failure and because either may compromise replication capacity. As additional NRTI mutations arose, replication capacity continued to drop, but more slowly than after emergence of the first NRTI mutation.
How different mutations affect boosted PIs
Analysis of 9860 HIV isolates shipped to Monogram for resistance testing yielded three noteworthy findings about resistance to ritonavir-boosted tipranavir and other PIs [7]:
- The I84V mutation--with or without other key PI mutations--had the biggest impact on all boosted PIs studied, including tipranavir.
- The common V82A mutation--without other critical PI mutations--had no affect on susceptibility to tipranavir and had little effect when joined by a second seminal PI mutation.
- L90M, another common PI mutation, had the least impact on resistance to any PI studied except saquinavir.
Peter Piliero of Boehringer Ingelheim, tipranavir's maker, cautioned that the analysis has limits. Most importantly, the proportion of viral isolates judged susceptible or resistant depends on the Monogram-defined upper clinical cutoff for resistance to tipranavir (8), amprenavir (11.5), saquinavir (12), atazanavir (20), and lopinavir (55). Statisticians controlled the analysis only for major PI mutations assessed in this study--any substitution at protease positions 23, 24, 30, 32, 46, 47, 48, 50, 54, 82 (except V82I), 84, 88, and 90, plus L33I and L33F. (V82I is a mutation selected by tipranavir.) The number of isolates with genotypic and phenotypic data differed for each of the PIs: 9860 isolates had genotypic and phenotypic data for amprenavir, lopinavir, and saquinavir, 9007 for atazanavir, and only 2272 for tipranavir. Finally, Piliero could not reckon susceptibility to a rival PI, darunavir (TMC114).
Nearly 60% of the isolates scrutinized had one or two key PI mutations, and 38.5% had two or three mutations. Two or more of the mutations studied rendered 50% of isolates resistant to amprenavir, 58% resistant to atazanavir, and 70% resistant to saquinavir. Three or more mutations made 70% of isolates resistant to lopinavir, 79% resistant to atazanavir, 82% resistant to saquinavir, and 85% resistant to amprenavir. In contrast only 17% of isolates with two key mutations and 38% with 3 key mutations proved fully resistant to tipranavir.
Table 2 shows the impact of various single and combined mutations on resistance to the PIs studied.
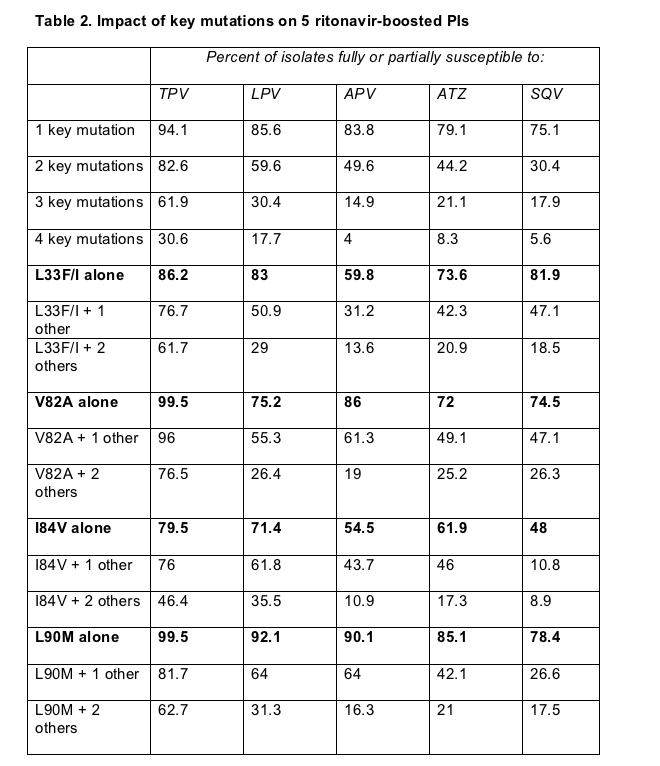
V82A, a mutation that arises with failing atazanavir, indinavir, lopinavir, ritonavir, or saquinavir, did not make HIV resistant to tipranavir, and rarely did so even when accompanied by one or two other key mutations. That makes sense, Piliero observed, because failure of tipranavir selects V82I or V82T, not V82A.
I84V alone had the greatest impact on every one of the PIs studied and, except for saquinavir, L90M had the least impact on these PIs. But L90M plus one other critical mutation made one third to three quarters of isolates resistant to lopinavir, amprenavir, atazanavir, and saquinavir, whereas L90M plus one made fewer than 20% of isolates resistant to tipranavir.
Just after ICAAC, Boehringer and a panel of experts published a list of 21 mutations at 16 protease positions that confer resistance to tipranavir (10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D, and 84V).[8] The scoring system that comes with this list calls virus with four or fewer of these mutations susceptible to tipranavir, virus with five to seven partially resistant, and virus with eight or more resistant.
Closer (clonal) looks show more mutations
Clonal analysis of viral populations from people taking a failing four-nucleoside regimen spotted mutations not seen by standard gene sequencing before treatment began and during virologic failure [9]. Researchers from GlaxoSmithKline and from sites of the COL40263 trial compared standard population sequencing results with clonal analyses in 14 people in whom once-daily Trizivir (AZT/3TC/abacavir) plus tenofovir failed. They defined failure as consecutive viral loads above 400 copies at or after treatment week 24. Pretreatment viral loads were higher and CD4 counts lower in the failure group, and a higher proportion of in the failure group were black (Table 3).
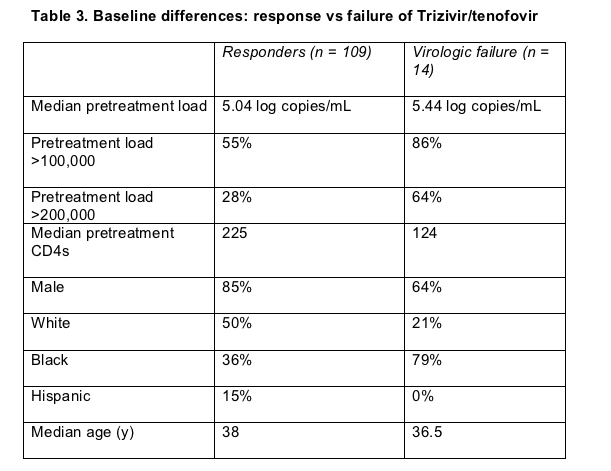
Standard genotyping saw NRTI and NNRTI mutations before treatment in 3 of 14 people who met failure criteria, while clonal analysis spotted pretreatment mutations in these 3 and in an additional 6. Seven of these 14 (4 with mutations detected only by clonal analysis) had pretreatment mutations related to study drugs. Additional thymidine analog mutations (TAMs) or K65R emerged in 6 of these 7 people during treatment with Trizivir/tenofovir.
Among 4 people in whom standard genotyping saw only nonmutant ("wild-type") virus before treatment and nonmutant virus upon failure, clonal analysis disclosed resistance-related mutations in 3 upon failure: L210W (a TAM), M184I (a 3TC mutation), and Y188L (a nonnucleoside mutation not related to drugs in this regimen). In the 10 people in whom standard population sequencing detected resistance mutations upon failure, clonal scrutiny invariably found additional nucleoside-related mutations.
Both standard sequencing and clonal analysis uncovered the tenofovir/abacavir-related K65R mutation when Trizivir/tenofovir stopped working in 2 people who had nonmutant virus or only the V118I substitution before treatment. (The significance of the V118I change in reverse transcriptase without V44D is unknown.[10]) Treatment with standard-dose AZT typically thwarts emergence of K65R, so the researchers speculated that the once-daily AZT dose used in this trial may explain why K65R popped up. Another possibility, they suggested, is that using abacavir with tenofovir allowed K65R to evolve. In 1 person in whom K65R emerged, clonal analysis detected a K70E substitution before therapy. Some research suggests that K70E may favor later selection of K65R.
The Glaxo researchers observed that their clonal analysis bolsters the earlier conclusion that TAMs--and specifically T215F instead of T215Y--are the primary route to resistance among people taking Trizivir with tenofovir. This resistance route differs from the M184V-plus-K65R pathway plotted when tenofovir-containing triple regimens without AZT fail [11-13].
How TAMs affect emergence of K65R
In people with no record of thymidine analog mutations (TAMs) when K65R emerged, TAMs never evolved along with K65R [14]. But this retrospective analysis of 51 people by Brenda Dauer (University of Frankfurt) found that a history of TAMs was a prerequisite for later coexistence of TAMs and K65R.
The cohort included 51 people in whom K65R emerged from 1999 through 2005 and who had genotyping results during treatment with regimens taken before K65R arose. The group included 34 people (67%) taking only NRTIs and 42 (82%) taking tenofovir. Among 30 people with no TAMs in their record, TAMs did not emerge along with K65R and never emerged as long as K65R remained detectable. Three of these people with no TAM history and no later TAMs were taking AZT, and 3 were taking d4T (the two thymidine nucleoside analogs).
Among 21 people with a history of TAMs before their current regimen, K65R alone evolved in 10 of them and K65R plus historical and additional TAMs arose in 11. Attempting to discern differences between these two groups, Dauer weighed the potential impact of previous regimens, AZT- and d4T-sparing regimens, treatment interruptions before the K65R-inducing regimen began, current mutation pathways, and TAM patterns just before the emergence of K65R. None of these variables helped explain why TAMs cropped up in some people, but not others, with K65R.
Dauer concluded that "a history of TAMs is a prerequisite for the coexistence of K65R plus TAMs, but not a predictor."
Resistance analysis of TDF/FTC-versus-AZT/3TC trial
After 96 weeks of follow-up in Gilead study 934, significantly more previously untreated people taking tenofovir/emtricitabine (TDF/FTC) than Combivir (AZT/3TC)--both with efavirenz--met the primary endpoint of a viral load below 400 copies in a time-to-loss-of-virologic-response analysis (75% versus 62%, P = 0.004) [15]. The better virologic response with TDF/FTC meant that significantly fewer people in that group met study criteria for resistance testing, and significantly fewer ended up with the M184V or M184I mutation, which evolves with failure of FTC or 3TC (Table 4) [16].
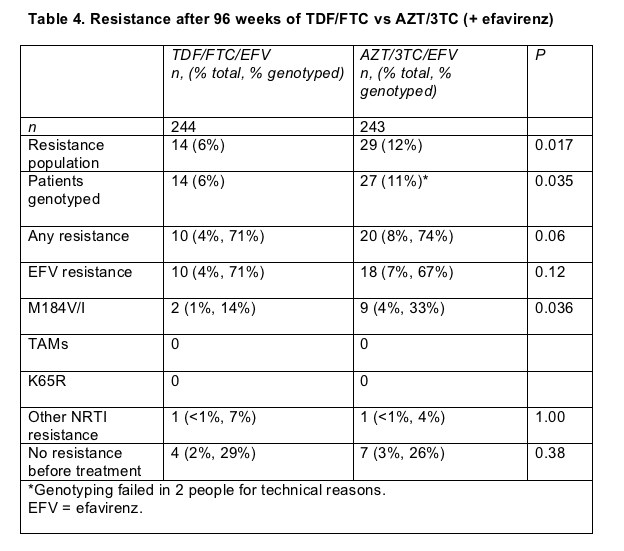
Failure of TDF/FTC/efavirenz evoked resistance to efavirenz at the same rate as failure of AZT/3TC/efavirenz did (71% versus 74%). M184V or M184I evolved less often than efavirenz-related mutations in both groups, but mutations at 184 arose more than twice as often upon failure of AZT/3TC than upon failure of TDF/FTC (33% versus 14%).
To learn more about the relative impact of FTC or 3TC on evolution of resistant virus, the Gilead team compared resistance rates among people taking TDF/FTC/efavirenz in this trial and treatment-naive people starting TDF/3TC/efavirenz in an earlier study [17]. Through 96 weeks of follow-up, significantly more people taking TDF/3TC than TDF/FTC required resistance testing (about 10% versus 5%, P = 0.018). Differences between those groups in proportions with resistant virus upon genotyping approached statistical significance (about 7% for TDF/3TC versus 4% for TDF/FTC, P = 0.061).
Among the 14 people with resistance mutations upon failure of TDF/FTC/tenofovir in the later trial [16], 1 had virus with decreased susceptibility to FTC and 3TC but not to NNRTIs, 1 had decreased susceptibility to FTC and 3TC and to efavirenz and nevirapine, and 6 had decreased susceptibility to efavirenz and/or nevirapine but not to NRTIs. This study also yielded evidence that people with HIV-1 subtypes other than subtype B respond to TDF/FTC/efavirenz as well as people with subtype B.
Adherence slips and viral blips
Do blips--those fleeting jumps from a sub-50 viral load into detectable territory--open the door to resistance? Three published reports involving a combined 154 people say they do [18-20], and one study of 10 people says they don't [21], although that last study divined a marginal correlation between blips and reported poor adherence (P = 0.08).
An ICAAC study by Abbott investigators also failed to link blips with evolution of resistant virus, even the hair-trigger M184V provoked by 3TC or FTC [22]. And blips did not presage virologic failure in this study, a result confirming most earlier reports. But the Abbott team did confirm the earlier suggestion [21] that adherence slips may cause blips. The findings deserve scrutiny because no one has nailed down what causes blips. Besides iffy adherence, viable explanations include dips in drug levels for other reasons [18], coinfection with another pathogen [20], a viral spill from activated latent reservoirs [18], or random biological or assay variation [21]. (For a review of studies addressing blips and resistance, see reference [23], which is free online. These Johns Hopkins authors maintain that the studies tying blips to resistance [18-20] are "complicated by limited assessment of pre-existing mutations as well as poor sensitivity of the genotyping assay employed.")
To learn whether wobbly adherence precedes blips, Abbott looked at virologic response and adherence records of treatment-naive people starting a first regimen containing either once- or twice-daily lopinavir/ritonavir plus tenofovir and emtricitabine (studies M99-956 and M02-418). Trial clinicians measured viral loads every 4 weeks through week 24, every 8 weeks through week 28, and every 12 weeks through week 96. Pill bottle caps fitted with microchips told researchers whether study participants opened their pill bottles at the right times.
The adherence study focused on 223 people with bottle-cap data, 131 taking once-daily lopinavir and 92 taking twice-daily lopinavir. Defining blips as a viral load spurt to between 50 and 1000 copies followed by a sub-50 load on the next reading, the researchers recorded blips in 60 people (27%), with no significant difference between those taking once-daily lopinavir (30%) and those taking the twice-daily dose (23%) (P = 0.212). The median blip measured 82 copies and ranged from 51 to 858 copies. Neither pretreatment viral load nor pretreatment CD4 count foretold blips. Blips did become more common as the studies proceeded, with a 23.3% blip frequency before week 24, 36.7% between week 24 and week 48, and 40.0% after week 48.
The Abbott team used bottle-opening data to reckon the average number of days a person took the prescribed doses in the week before a blip--and the average number of days with correct adherence during a week when the same person had no blips. The average for the pre-blip period proved significantly lower than the no-blip average (5.55 versus 6.22 days, P = 0.007). Bottle-cap data also recorded a jump in correct adherence just after most blips. The trial design mandated a follow-up viral load reading within 4 weeks of any blip. When people learned their viral load spike was a blip and not a full-fledged rebound, adherence subsided from this higher post-blip level back to the pre-blip plateau.
Mark Mascolini writes about HIV infection (markmascolini@earthlink.net).
References
1. Ross L, Florance A, Wine B, et al. Factors associated with HIV-1 mutations to drug resistance in antiretroviral therapy naive HIV-infected individuals in the United States from 2000-2005 (PREPARE Study). 46th ICAAC. September 27-30, 2006. San Francisco. Abstract A-378.
2. Little SJ, May S, Hecht F, et al. Increase in transmitted NNRTI drug resistance among recently HIV-infected patients from North America and Australia. Antiviral Ther 2006;11:S110.
3. Smith D, Pesano R, Cachay E, et al. Prevalence of HIV drug resistance among antiretroviral naive individuals of unknown infection duration. Antiviral Ther 2006;11:S121
4. Eshelman SH, Husnik M, Hudelson S, et al. Analysis of antiretroviral drug resistance and HIV-1 subtype among men who have sex with men recently infected with HIV-1 in the United States: the EXPLORE Study. Antiviral Ther 2006;11:S122.
5. Shet A, Berry L, Mohri H, et al. Tracking the prevalence of transmitted antiretroviral drug-resistant HIV-1: a decade of experience. JAIDS 2006;41:439-446.
6. Ross L, Parkin N, Lanier R. The number of HIV primary NRTI mutations correlates directly with other antiretroviral associated mutations and indirectly with replicative capacity and reduced drug susceptibility. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1001.
7. Piliero PJ, Narkin N, Mayers D. Impact of protease mutations L33F/I, V82A, I84V, and L90M on ritonavir-boosted protease inhibitor susceptibility. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-998.
8. Baxter JD, Schapiro JM, Boucher CA, et al. Genotypic changes in human immunodeficiency virus-1 protease associated with reduced susceptibility and virologic response to the protease inhibitor tipranavir. Virology 2006;80:10794-10801.
9. Rouse E, Preble L, Elion R, et al. Comparison of clonal genotyping and population genotyping of HIV reverse transcriptase at baseline and virologic failure in patients on once-daily Trizivir and tenofovir for <48 weeks. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1005.
10. Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: 2005. Topics HIV Med 2005;13:51-57.
11. Khanlou H, Yeh V, Guyer V, Farthing C. Early virologic failure in a pilot study evaluating the efficacy of therapy containing once-daily abacavir, lamivudine, and tenofovir DF in treatment-naive HIV-infected patients. AIDS Patient Care STDs 2005;19:135-140.
12. Gallant JE, Rodriguez AE, Weinberg WG, et al. Early virologic nonresponse to tenofovir, abacavir, and lamivudine in HIV-infected antiretroviral-naive subjects. J Infect Dis 2005;192:1921-1930.
13. Jemsek J, Hutcherson P, Harper E. Poor virologic responses and early emergence of resistance in treatment naive, HIV-infected patients receiving a once daily triple nucleoside regimen of didanosine, lamivudine, and tenofovir DF. 11th Conference of Retroviruses and Opportunistic Infections. February 8-11, 2004. San Francisco. Abstract 51.
14. Dauer B, Staszewski S, Babacan E, et al. History of TAMs is a prerequisite for co-existence of the K65R mutation plus TAMs but is not a predictor. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1002.
15. Gallant J, Pozniak A, DeJesus E, et al. Efficacy and safety of tenofovir DF, emtricitabine and efavirenz (EFV) compared to fixed dose zidovudine/lamivudine and EFV through 96 weeks in antiretroviral treatment-naive patients. XVI International AIDS Conference. August 13-18, 2006. Toronto. Abstract TUPE0064.
16. McColl DJ, Margot N, Chuang S, et al. Study 934: lower rates of resistance development associated with tenofovir DF and emtricitabine plus efavirenz at week 96. 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-1004.
17. Gallant JE, Staszewski S, Pozniak AL, et al. Efficacy and safety of tenofovir DF vs stavudine in combination therapy in antiretroviral-naive patients: a 3-year randomized trial. JAMA 2004;292:191-201.
18. Macias J, Palomares JC, Mira JA, et al. Transient rebounds of HIV plasma viremia are associated with the emergence of drug resistance mutations in patients on highly active antiretroviral therapy. J Infect 2005;51:195-200.
19. Cohen Stuart JW, Wensing AM, Kovacs C, et al. Transient relapses ("blips") of plasma HIV RNA levels during HAART are associated with drug resistance. JAIDS 2001;28:105-113.
20. Easterbrook PJ, Ives N, Waters A, et al. The natural history and clinical significance of intermittent viraemia in patients with initial viral suppression to <400 copies/ml. AIDS 2002;16:1521-1527.
21. Nettles RE, Kieffer TL, Kwon P, et al. Intermittent HIV-1 viremia (blips) and drug resistance in patients receiving HAART. JAMA 2005;293:817-829.
22. Podsadecki TJ, Vrijens B, Tousset E, et al. Lower adherence to HAART observed prior to transient HIV-1 viremia ("blips"). 46th ICAAC. September 27-30, 2006. San Francisco. Abstract H-992.
23. Lee PK, Kieffer TL, Siliciano RF, Nettles RE. HIV-1 viral load blips are of limited clinical significance. J Antimicrob Chemother 2006;57:803-805 (http://jac.oxfordjournals.org/cgi/content/full/57/5/803).
|
| |
|
|
|
|
|