 |
|
|
| |
The HIV-1 Protease Inhibitor PL-100 Has a High Genetic Barrier and Selects a Novel Pattern of Mutations
|
| |
| |
Reported by Jules Levin
15th Intl HIV Drug Resistance Workshop
June 13-17, 2006, Sitges, Spain
J.J. Wu1, S. Dandache1, B. Stranix1, C. Panchal1, and M.A. Wainberg21
Ambrilia Biopharma Inc. and 2McGill University, Montreal, Canada
Author Conclusions
- The data show that there is a high genetic barrier to the development of resistance toPL-100, since four mutations in PR and potential additional mutations in Gag cleavage cleavage sites need to accumulate in the viral genome to observe only a mild resistance to our drug. In addition, the unique P81S mutation was shown to be lethal to the virus.
- The mutations selected by PL-100 do not confer any cross resistance in vitro to other PIs. In contrast, T80I induces hypersensitivity to SQV and NFV.
- The high genetic barrier and favorable PK profile suggest that PPL-100 has the high potential as a first-line QD PI.
Abstract
BACKGROUND:PL-100 is a novel HIV-1 protease inhibitor (PI) with a favorable cross- resistance profile. Its phosphorylated pro-drug PPL-100, with significantly improved solubility and pharma- cokinetics, has potential as a QD PI and is currently in Phase I human clinical trials.
METHODS: Mononuclear cells infected with laboratory-adapted HIV-1 strain IIIB were subjected to increasing concentrations of PL-100 or amprenavir. Sequencing of the protease (PR) region of viruses was performed using Bayer's HIV genotyping test. Quickchange site-directed mutagenesis kit was used to introduce identified PR mutations into pNL-4.3. Phenotyping of produced viral mutants was performed using MT-4 cytoprotection assay (MTT). Standard viral growth studies were conducted in MT-4 cells to analyze viral replication capacity.
RESULTS: While amprenavir selected for the expected signature mutations, after 48 weeks of passaging under increasing selective pressure of PL-100, we observed a novel pattern of mutations (K45R, M46I, T80I, and P81S) in the PR gene. T80I was observed at week 8. All other mutations did not appear until week 25. No further mutations were observed up to 48 weeks. K45R and M46I are known mutations in the flap region; T80I and P81S are novel mutations in the active site. Site-directed mutagenesis reveals that P81S is a lethal mutation, since the replication capacity of various mutants containing this mutation was severely impaired except the mutant containing all four mutations (K45R, M46I, T80I and P81S). These observations correlated well with loss of PR activity in the P81S mutants and recovery of proteolytic activity when all four mutations present, as measured by p55 Gag processing. Single, double or triple viral mutants did not show resistance to PL-100 (EC50 fold-change (FC) < 2.5), while only mild resistance to PL-100 was observed with the quadruple viral mutant (FC = 10.8). No cross-resistance to amprenavir, lopinavir, atazanavir, saquinavir, indinavir, and nelfinavir was observed (FC < 2.5) for any viral mutants. Interestingly, T80I induced hypersensitivity (22-fold) to saquinavir. Finally, pharmacokinetic data suggest that PPL-100 is likely a QD PI without RTV boosting.
CONCLUSION: The novel mutational pathway and favorable pharmacokinetic profile justify further clinical development of the drug to treat PI-naive and experienced patients.
Introduction
The development of new HIV inhibitors with distinct resistance profiles is paramount to derailing the emergence of multi-resistant strains. Our drug discovery program integrated viral resistance into the screening process of candidate molecules and produced PL-100, a novel and potent Protease Inhibitor (PI). It was previously shown that this novel PI had a favorable cross-resistance profile when tested on a panel of 63 strains representing isolates present in highly PI experienced HIV+ patients. Its phosphorylated pro-drug (PPL-100), with significantly improved solubility and pharmacokinetics, has the high potential as a QD PI and is currently in Phase I human clinical trials. Recently, a 48-week in vitro resistance selection study was performed to determine the degree of difficulty for HIV to develop resistance to PL-100.
Materials and Methods
For the in vitro selection of HIV variants able to grow in presence of PL-100, Blood Mononuclear Cells (BMCs) (2-4 x106 cells) were infected with the laboratory- adapted strain HIV/IIIb at a multiplicity of infection of 1 (moi=1) in presence of suboptimal concentrations of PL-100 or Amprenavir (APV) as a control. Fresh donor CBMCs were added weekly and the concentration of the compounds was increased 2 to 2.5-fold at each passage unless the pressure on viral survival was too great. At each passage, an aliquot of the supernatant was taken and virus concentration was determined by RT. Viral genotyping was performed using the TRUGENE HIV Genotyping Kit (Bayer) The PR mutations identified by sequencing were introduced in pNL-4.3 by the Quickchange Site-Directed Mutagenesis kit following the manufacturer's protocol (Stratagene). Viral stocks were produced in MT4 or COS-7 cells by transfection and quantified by p24 ELISA ((Vironostika HIV-1 Antigen, Microlesia System, bioM_rieux). The in vitro susceptibility of HIV-1 NL-4.3-based site-directed mutants to PL-100 and currently marketed PIs (except Ritonavir) was determined by a MTT cytoprotection assay measuring the inhibition of the cytopathic effects induced by HIV-1 in MT-4 cells.For the viral Gag precursor protein processing analysis, COS-7 cells were transfected with NL-4.3-based proviral clones by the Calcium-phosphate method. 48 hours post-transfection, cells were lysed and aliquots of equal volumes were loaded on a 12% NuPAGE BIS-Tris gel, followed by western blotting using an anti-HIV-1 p24 monoclonal antibody. The band intensities on film were then measured using ImageQuant 5.0 (Molecular Dynamics)The replication capacity of the SDMs was evaluated using standard viral growth kinetics in MT4 cells. Equal amounts of each SDM (18ng of p24) were used to infect MT 4 cells (0.6x106) that were then cultured for seven days. Viral replication was measured by p24 ELISA at the time points indicated.
RESULTS & DISCUSSION
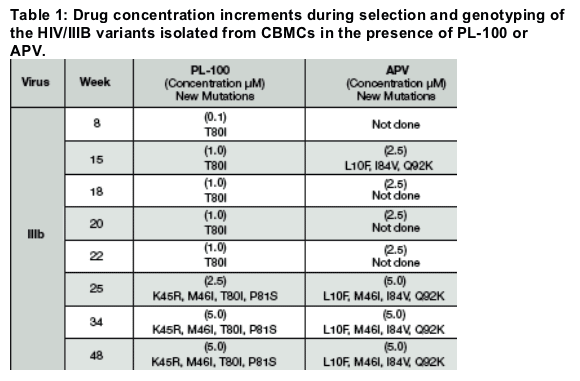
The genotyping of the virus growing in the presence of APV revealed expected mutations in HIV-1 Protease (PR).However, HIV-1/IIIb variants able to grow in the presence of PL-100 harbored several unique mutations in the PR region of their genome. First, at week 8 appeared the T80I mutation that was maintained throughout the 48 week selection. When the PL-100 concentration was increased, a quadruple mutant was selected with K45R, M46I and P81S in addition to the original T80I mutation. Interestingly, we were never able to isolate a virus with an intermediate genotype between the single mutant and the quadruple mutant.
Mutation in K45R, although not currently considered a resistance mutation, has been recently associated with Nelfinavir treatment, in patients that failed first-line regimen containing NFV. In this context, this novel mutation clustered only withD30N and N88D, two NFV-resistance mutations. It is very rare in PI-naive patients.
M46I is a well-known flap tip mutation that seems to strongly influence the dynamics of flap opening. It was shown in co-crystal with IDV and RTV to change the conformation of the 80's loop. It is thought to be a true compensatory mutation. M46I is known to be present in strains that are cross-resistant to several PIs.
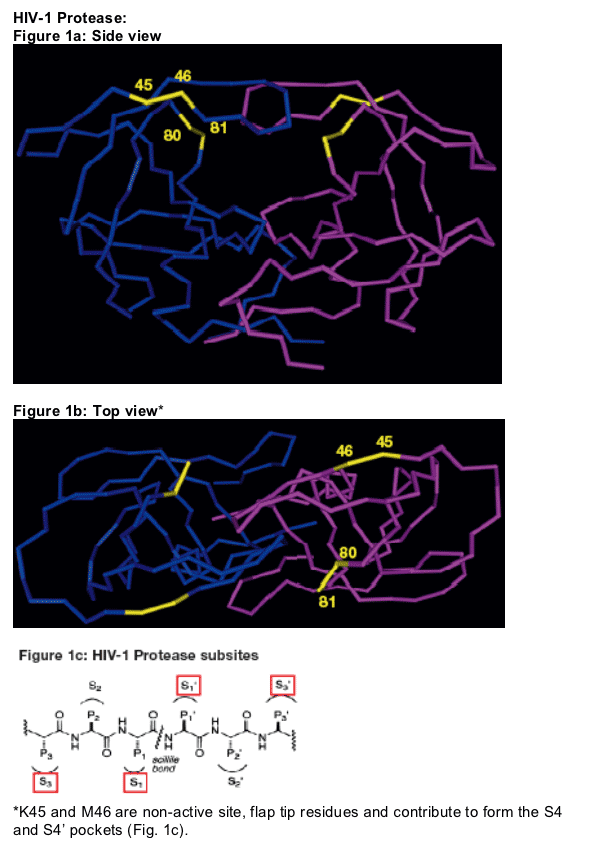
*T80 and P81 are substrate cleft residues and are both highly conserved in HIV-1, HIV-2 and SIV. They are located in S1 and S1' pockets (Fig.1c), thus interacting with the P1 and P1' side chains of the substrate/inhibitor. P81 also contributes to formation of the S3 and S3' pockets (Fig.1c).
Both T80I and P81S are previously un-described, non-conservative mutations.
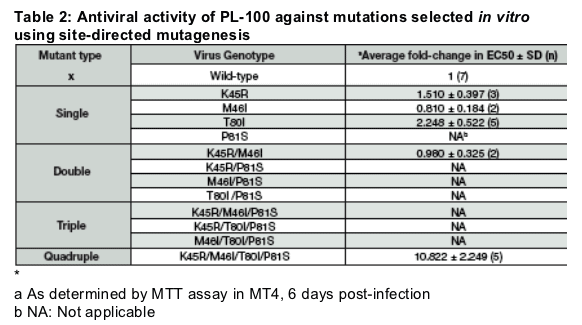
The PR mutations selected under PL-100 pressure were introduced in the HIV-1 laboratory-adapted proviral clone pNL-4.3 by site-directed mutagenesis (SDM). PL-100's antiviral activity against the generated SDM viruses was measured by MTT assay in MT4 cells.
The P81S-containing mutants were all defective and could not be tested except for the quadruple mutant that conferred only a mild resistance to PL-100. This suggests that PL-100 is a high genetic barrier drug.
We are currently sequencing the Gag protease cleavage sites of the selected variants to determine whether mutations in these secondary sites are involved in the ability of the virus to grow in presence of PL-100.
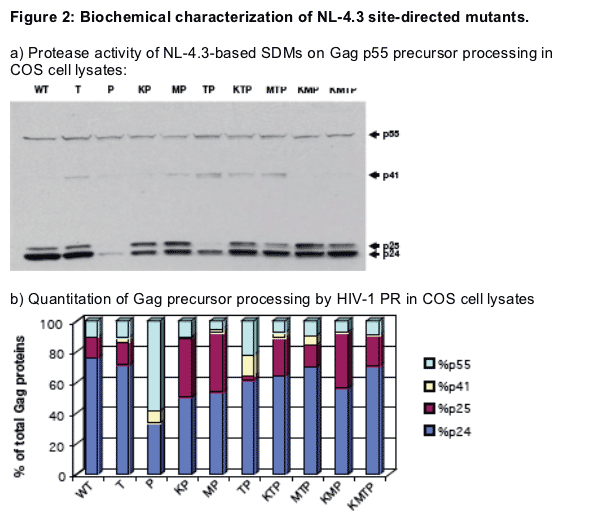
HIV-1 Gag processing was studied in COS cells transfected with NL-4.3 site-directed mutants. Western blotting of cell lysates with an HIV-1 p24 monoclonal antibody was followed by quantitation of band intensities.
Mutants K45R, M46I and T80I had only a minor defect in Gag processing. However, mutation P81S was shown to affect very significantly the Gag processing, suggesting a major defect in HIV-1 Protease in the context of this mutant.
Only when the P81S mutation is in the context of the quadruple mutant, do we observe a polyprotein processing close to that of the wild-type. P81S-based double mutants were all shown to be defective.
The analysis of triple and quadruple mutants suggest that mutations K45R and M46I are both required to compensate for the PR enzymatic defect caused by the P81S mutation.
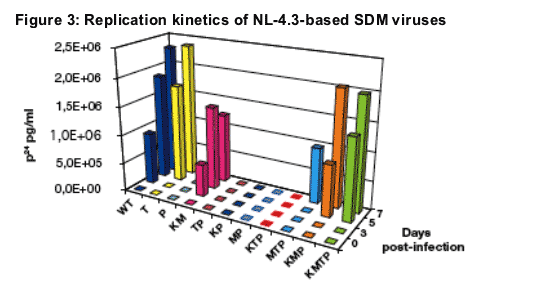
The analysis of the replication capacity of the NL-4.3-based SDM viruses in standard growth kinetics in MT4 revealed that there is a perfect correlation between the extent of Gag polyprotein processing and the replication capacity of the SDM viruses.
Indeed, P81S single and double SDMs were not infectious.
The analysis of P81S triple mutants allows to establish a hierarchy in the compensatory effects of the flap mutations: M46I>K45R.
The quadruple mutant, although slightly delayed compared to wild-type, had a replication capacity that was similar to the one of the T80I single mutant.
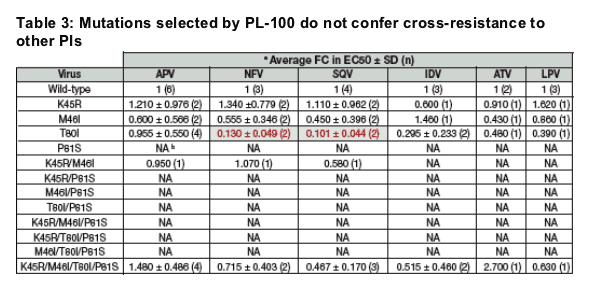
The phenotyping of the NL4.3 SDMs against all FDA-approved PIs (except RTV) revealed that the selected mutations did not confer any cross-resistance to the commercially available PIs.
Interestingly, the T80I single mutant was shown to confer hyper-susceptibility to SQV and NFV (~10-fold and ~8-fold increase in susceptibility, respectively).
|
| |
|
|
|
|
|