 |
|
|
| |
Interim analysis: results from a phase 2 study of telaprevir with peginterferon alfa-2a and ribavirin in treatment-naive subjects with hepatitis C: Prove 1
|
| |
| |
Reported by Jules Levin
AASLD, Nov 2-6, 2007, Boston, MA
Ira Jacobson, Gregory T Everson, Stuart C Gordon, Robert Kauffman, Lindsey McNair, Andrew Muir and John McHutchison on behalf of the PROVE1 study team
AUTHOR SUMMARY
61% SVR rate in the 24 week TVR based treatment group:
--RVR in 79% of TVR-treated subjects
--relapse rate was 2.4% in subjects with RVR who completed 24 weeks of treatment.
End of treatment (EOT) response rate was 45% in the 48-week control arm, and 65% in the 48-week TVR arm
The most common adverse events were those that are expected with Peg-IFN and RBV:
--Skin adverse events (rash, pruritis) were more common and more severe, and gastrointestinal events and anemia were more common, in the TVR groups.
AUTHOR CONCLUSIONS
Telaprevir based therapy drives-
--high rate of RVR and subsequent on-treatment response rates
--low rate of relapse
These results suggest the potential for higher SVR rates than currently expected with standard of care, with shortened duration of therapy.
Larger studies are required to confirm the safety and efficacy of this novel approach to HCV therapy.
PROVE1 study design
Phase 2, 250-subject, randomized, placebo-controlled trial conducted in the USA
Genotype 1 infected, treatment-naive patients
Standard inclusion/exclusion criteria for Peg-IFN/RBV studies; no cirrhosis
Study drug dosing:
--telaprevir (TVR) 750 mg q8h or placebo
--peginterferon alfa-2a 180ug SC weekly
--ribavirin 1000/1200 mg daily
Roche Taqman HCV RNA assaY-limit of detection 10 IU/ml
PROVE1 STUDY DESIGN
Patients receive 12 weeks of triple therapy. Arm b receives 36 additional weeks of peg/rbv. Arm c receives only 12 weeks of additional peg/rbv. Arm d only receives 12 weeks of triple (n=17).
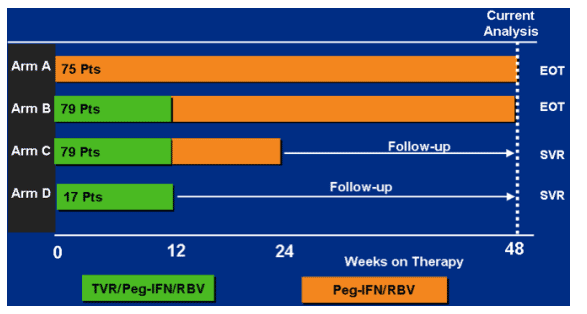
subjects in arms c and d needed an RVR at week 4, and had to remain HCV RNA undetectable through end of assigned dosing, to end treatment at week 24 and 12.
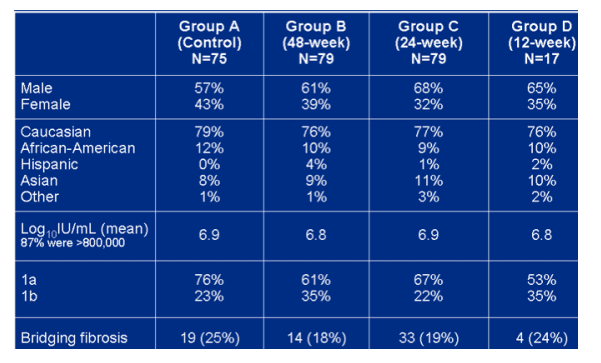
Baseline demographics
Viral load is 6.9 logs across all groups. 18-25% had bridging fibrosis. 9-12% are African-American.
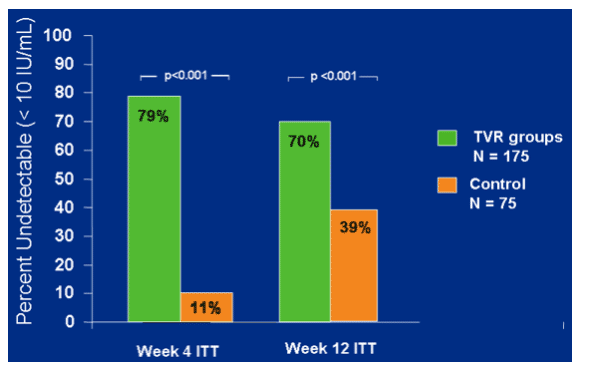
TVR groups had significantly better antiviral response at week 4 and week 12.
At week 4 79% in TVR groups and 11% in control had <10 IU/ml; at week 12 70% in TVR groups and 39% in control had <10 IU/ml. ITT (p<0.001)
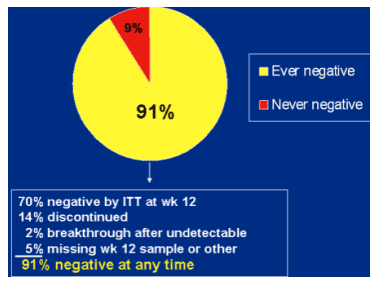
Patients with any undetectable HCV RNA on telaprevir d=uring first 12 weeks.
70% negative by ITT at week 12.
14% discontinued.
2% breakthrough after undetectable
5% missing week 12 sample or other
91% negative at any time.
Viral breakthrough - first 12 weeks
Increase in HCV RNA during first 12 weeks, either:
-- >100 IU/ml after becomingf undetectable
-- 1-log HCV RNA increase from nadir
fourteen subjects experienced viral breakthrough
2 in control group (3%)
12 in TVR groups (7%)
- all but one breakthrough in the TVR groups occurred within the first 4 weeks
- 9 of 12 breakthroughs occurred in patients who never became undetectable
- all associated with high level TVR-resistant viral variants
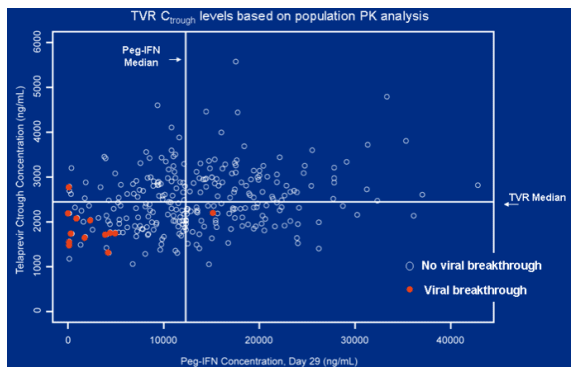
Viral breakthrough was associated with lower levels of peginterferon and TVR at day 29. Note from Jules: maybe ritonavir boosting could improve this and improve response rates.
SVR rates for groups C and D ITT Analysis
Group C (TVR/P/R 12 week, P/R 12 wk): 61%. 10% (n=8) lost to followupo, outcome not known, undetectable at last assessment.
--group D (TVR/P/R 12 week): 35%; 6% (n=1) lost to followup, outcome not known, undetectable at last assessment.
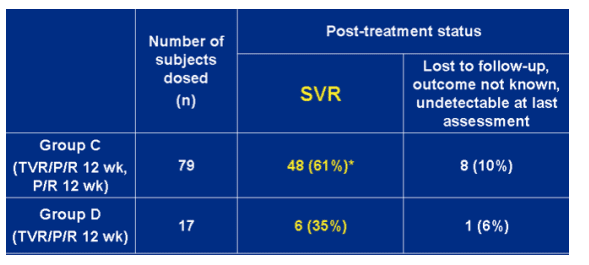
*includes 41 subjects who completed 24-week treatment and achieved SVR, and 7 subjects who discontinued early and achieved SVR.
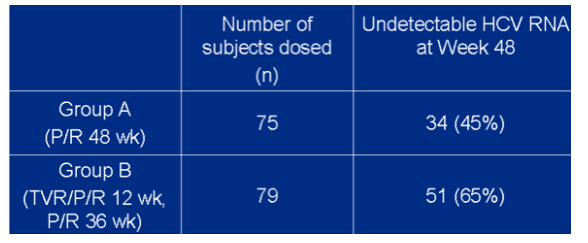
End of treatment response in 48-week arms ITT analysis
--group A (P/R 48 wk): 45% (n=34) undetectable HCV RNA
--group B (TVR/P/R 12 wk, P/R 36 wk): 65% (n=51)
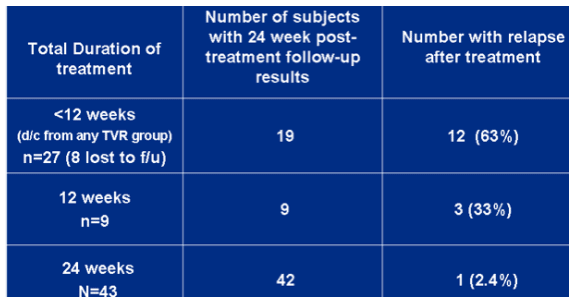
Relapse rates in subjects with undetectable HCV RNA at the end of TVR-based treatment.
<12 weeks (n=19), 63% relapsed. 12 weeks (n=9), 33% (n=3) relapsed. 24 weeks (n=43), 2.4% (n=1) relapsed.
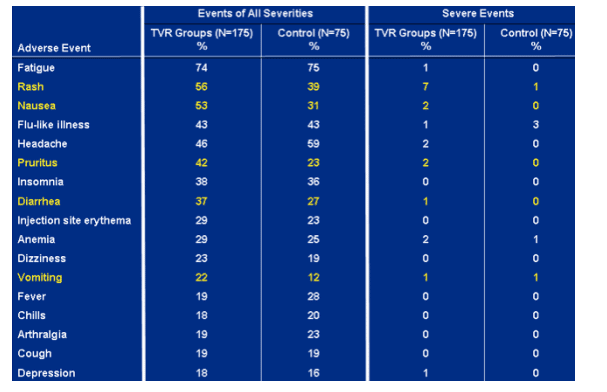
Most common adverse events (through week 48).
Rash: events of all severity- 56% TVR groups; 39% control; severe events- 7% TVR groups, 1% control.
Nausea: events of all severity- 53% TVR groups, 31% control; severe events- 2% TVR groups, 0 control.
Pruritis: events of all severity- 42% TVR groups, 23% control; severe events- TVR groups 2%, 0 control.
Diarrhea: events of all severity- 37% TVR groups, 27% control; severe events- 1% TVR groups, 0 control.
Vomiting: events of all severity- 22% TVR groups, 12% control; severe events- 1% in each group.
RASH: maximum severity
Any time on treatment
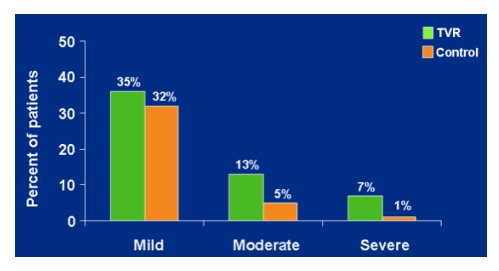
mild- 35% TVR, 32% control.
Moderate- 13% TVR, 5% control.
Severe- 7% TVR, 1% control.
Median time to onset of severe rash was 55 days
--investigators report that rashes resolve on discontinuation of study drug.
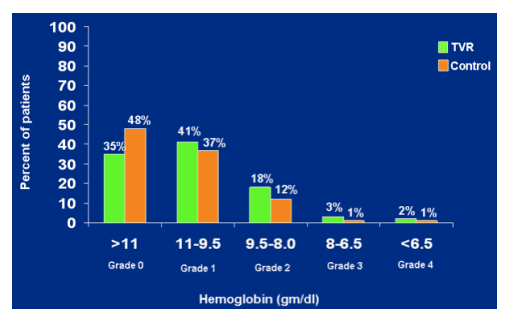
Hemoglobin: nadir value
Any time on treatment
Grade 1 11-9.5 gm/dl: 41% TVR, 37% control; grade 2 8-6.5: 3% TVR, 1% control; <6.5 grade 4: 2% TVR, 1% control.
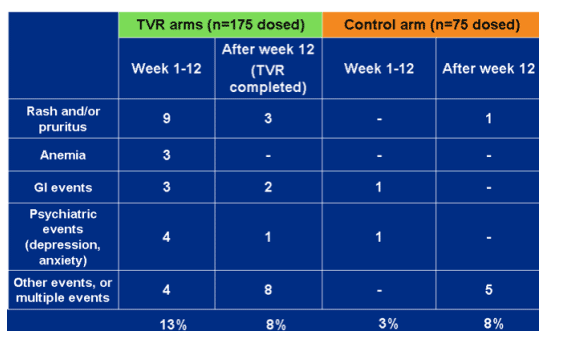
Adverse events leading to discontinuation through week 48.
Total 13% on TVR during week 1-12, 8% after week 12. On control, 3% week 1-12, 8% after week 12. Events included rash/pruritis, anemia, GI, psych, other.
|
| |
|
|
|
|
|