 |
|
|
| |
Abbott Reports 4 Viral Decay Phases: Low-Level Viremia Decays Over 7+ Years to a Residual Level Correlated with Baseline HIV-1 RNA
|
| |
| |
Reported by Jules Levin
CROI, Feb 2007, Los Angeles
M King1, S Palmer2, A Wiegand2, F Maldarelli2, B Bernstein1, D Kempf1, GJ Hanna1, J Mellors3, J Coffin2
1Abbott, Abbott Park, IL; 2HIV Drug Resistance Program, NCI, NIH, Bethesda, MD; 3University of Pittsburgh, Pittsburgh, PA
AUTHOR DISCUSSION
We observed a non-linear decline in plasma HIV-1 RNA below 50 copies/mL beginning 1 year after initiation of suppressive therapy, suggesting additional phases of viral dynamics beyond the previously described 1st and 2nd phases of decay.
Initial analyses fitting a model with two distinct decay phases could not detect a decay in the latter phase. Thus, the model used for the analysis replaced this term with a constant term to reflect a decay rate of zero.
Biologically, our model therefore implies that low-level persistent viremia arises from two compartments, one from which viral production decays over time and a second from which viral production remains stable.
Results of the fitted model indicated that the former compartment typically accounts for baseline viremia of approximately 5-6 copies/mL and decays with a half-life of approximately 15 months, while the latter compartment typically accounts for viremia of approximately 1 copy/mL, with no decay.
Statistically significant correlations between subject-specific estimates of the pre-treatment viremia in each compartment and total baseline viral load were observed in each study (even though the model used only data from >1 year after treatment initiation), supporting a link between the extent of infection before therapy and the size of long-lived viral reservoirs.
A key limitation of our results is imposed by the simplicity of our model, which allows for only two compartments, one of which decays and one of which remains stable over time. Either of the "compartments" we specify could represent multiple biological phenomena.
Furthermore, the "stable" compartment in our model may decay at a rate too slow to be detected in our study.
CONCLUSIONS
We observed a non-linear decline in plasma HIV-1 RNA below 50 copies/mL beginning 1 year after initiation of suppressive therapy, suggesting additional phases of viral dynamics beyond the relatively rapidly decaying first two phases.
A third-phase half-life of 14-15 months was observed in each of the two studies, and no decay during a fourth phase was discernable.
Low-level persistent viremia appears to arise from at least two cellular compartments, one with a half-life comparable to that previously described for latently infected CD4+ T cells (6-44 months), and one or more with a half-life too long to be detected over the 7-year span of our data.
Subject-specific estimates of baseline viremia from each of the two compartments were correlated with total baseline viremia, supporting the hypothesis that persistent viremia over time comes from sources or compartments that were present prior to initial treatment.
BACKGROUND
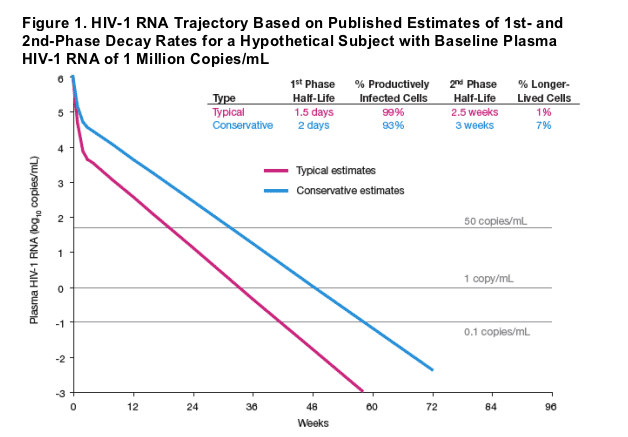
Antiretroviral treatment of HIV-1 infection results in an initial biphasic decay in plasma HIV-1 RNA, representing rapid clearance (half-life of 1-2 days) of virus from short-lived infected cells (1st phase) and more gradual clearance (half-life of 2-3 weeks) from longer-lived cells (2nd phase) (Perelson 1997, Wu and Ding 1999, Haubrich 2007). Plasma HIV-1 RNA trajectories predicted by these studies, displayed in Figure 1, suggest that after 60 weeks of treatment, residual viremia from the first 2 phases of decay for a subject with baseline plasma HIV-1 RNA of 1 million copies/mL plasma would be <0.001 to 0.07 copies/mL. However, residual plasma HIV-1 RNA (2-4 orders of magnitude higher than these predicted levels) persists beyond 60 weeks of treatment (Palmer 2006), suggesting additional compartments expressing HIV-1. The dynamics of plasma viral load beyond the 1st and 2nd phases have not been well-described, due in part to the lower limit of quantitation for readily available HIV-1 RNA assays and limited availability of samples from subjects with consistent long-term suppression of HIV-1 RNA. In the current study, we retrospectively applied a highly sensitive assay to samples from treatment responders in two long-term clinical studies of protease inhibitor-based therapy.
METHODS
Longitudinal plasma samples were obtained from two studies from treated subjects who consistently had plasma HIV-1 RNA <50 copies/mL by conventional assays.
- Study 720: 293 samples from 40 subjects receiving lopinavir/ritonavir (LPV/r) plus stavudine [d4T] and lamivudine [3TC]. Samples collected between Week 60 and Week 360 (Year 7) were analyzed.
- Study 863: 370 samples from 117 subjects receiving LPV/r or nelfinavir, plus d4T and 3TC. Samples collected between Week 60 and Week 110 were analyzed.
Plasma HIV-1 RNA was quantitated using a previously described HIV-1 RNA assay with single copy sensitivity (Palmer 2003). Based on sample volumes available, the assay detection limit was typically 0.4-0.7 copies/mL.
Using nonlinear mixed effects models, we assessed HIV-1 RNA decay from Week 60 through 7 years of treatment. These models allowed for up to two distinct phases of decay, representing viremia arising from a 3rd and 4th compartment beyond the first 2 phases of decay previously published.
Model 1: V(t) = V3i exp(-_it) + V4i exp(-_t)
Model 2: V(t) = V3i exp(-_it) + V4i
Parameters of these models are interpreted as follows:
V(t) = plasma viral load at time t
i = 1 (for Study 720) or 2 (for Study 863)
V3i = baseline viral load in compartment 3 for study i
_i = decay parameter for compartment 3 in study i
V4i = baseline viral load in compartment 4 for study i
_ = decay parameter for compartment 4 (Model 1 only)
Notably, Model 1 allows for decay of compartment 3 and compartment 4 each, while Model 2 allows decay of compartment 3, but considers compartment 4 to remain stable (no decay).
Empirical Bayes estimates of subject-specific baseline viremia in each compartment were compared to total baseline viremia (Week 0) in each study using linear regression.
RESULTS
Initial fits of Model 1 did not detect any decay associated with compartment 4 (p=0.98 for the null hypothesis of no decay), implying the half-life of compartment 4 could not be detected over the 7-year range of our data. Therefore, subsequent analysis was based on Model 2, which assumes compartment 4 does not decay over time.
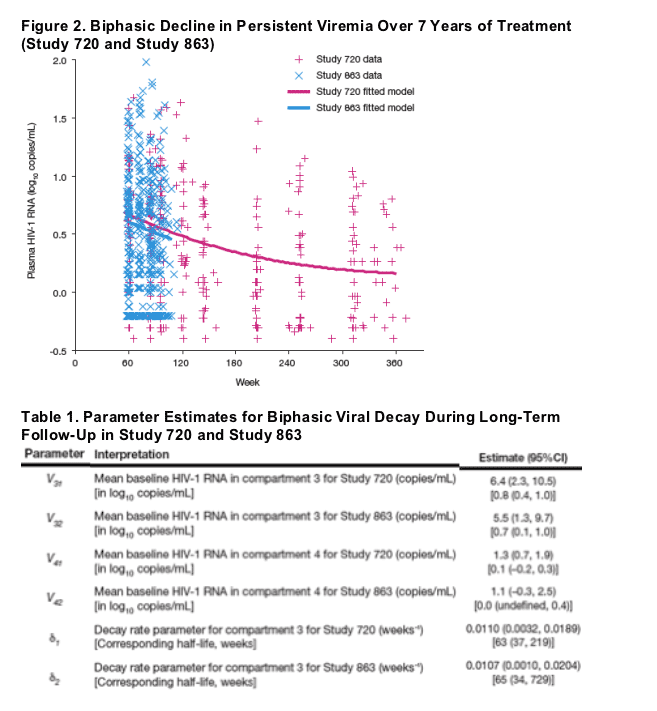
Based on Model 2, we observed a statistically significant decline in HIV-1 RNA after Week 60 that was consistent between studies (p=0.91 for the difference between studies). The fitted model provided half-life estimates for compartment 3, representing a third phase of viral load decay, of 63 weeks for Study 720 and 65 weeks for Study 863.
By years 5-6, residual viremia began to level off, converging to the model-specified fourth phase of low-level plasma viremia with no detectable decay over time (Figure 2).
Estimates of contribution of the third and fourth compartments to overall baseline viral load were <0.1% (6-8 copies/mL).
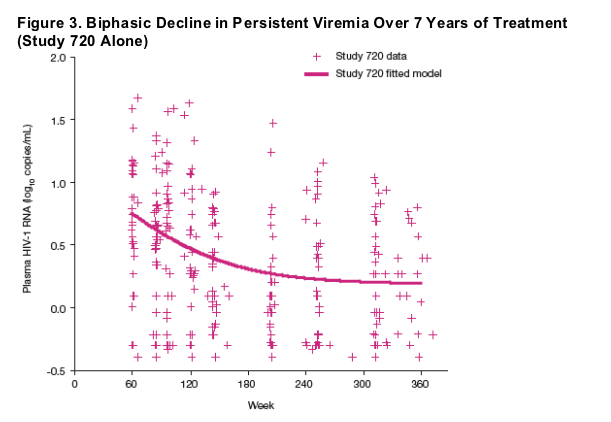
Results were comparable if only the data from Study 720 were used to fit Model 2 (Figure 3), providing a half-life of 39 weeks (95% CI, 25 to 90 weeks) and a mean residual persistent viremia level (compartment 4) of 1.5 copies/mL.
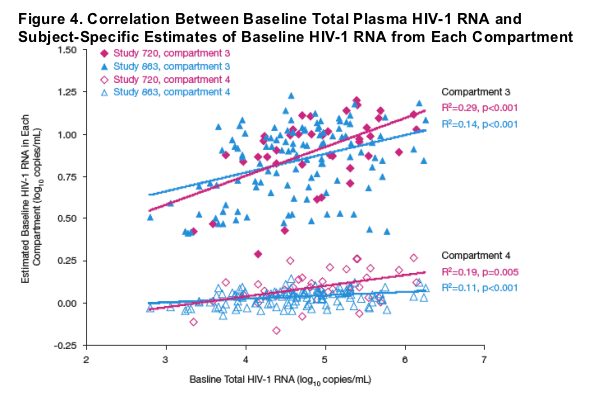
Empirical Bayes estimates of baseline HIV-1 RNA from compartment 3 and compartment 4 each were statistically significantly correlated with baseline viremia (Figure 4).
- Compartment 4: Study 720, R2=0.19, p=0.005; Study 863, R2=0.11, p<0.001
- Compartment 3: Study 720, R2=0.29, p<0.001; Study 863, R2=0.14, p<0.001.
References
Haubrich R, et al. 14th CROI, Los Angeles, 2007.
Palmer S, et al. J Clin Microbiol. 2003;41:4531-6.
Palmer S, et al. 15th HIV Drug Resistance Workshop, Sitges, Spain, 2006. Abstract 55.
Perelson AS, et al. Nature. 1997:387:188-91.
Wu H, Ding AA. Biometrics. 1999;55:410-8.
|
| |
|
|
|
|
|