 |
|
|
| |
Interim Results Presented at EASL from PROVE 1 Clinical Trial of Investigational Drug Telaprevir in Patients with Genotype 1 Hepatitis C
|
| |
| |
PROVE 1 data support potential to shorten treatment duration in treatment-naive, genotype 1 HCV patients
Press Release from Vertex
Barcelona, Spain, April 14, 2007 - In a late-breaker presentation at the 42nd Annual Meeting of the European Association for the Study of the Liver (EASL), researchers today presented data from a planned interim analysis of the PROVE 1 clinical trial, which is the first trial to evaluate short-duration therapy with the investigational hepatitis C protease inhibitor telaprevir (TVR, VX-950) in combination with pegylated interferon (peg-IFN) and ribavirin (RBV) in treatment-naive, genotype 1-infected hepatitis C patients. The data from PROVE 1 demonstrated a high rate of rapid viral response (RVR) in the telaprevir groups and a low rate of on-treatment viral breakthrough, and suggested that 12 weeks of telaprevir-based therapy enabled some patients to clear the virus. Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) is developing telaprevir in collaboration with Tibotec.
"The high rates of RVR observed in the telaprevir groups in PROVE 1, and the fact that some patients have remained persistently viral negative 20 weeks after stopping the 12 weeks of telaprevir-based therapy, suggest that we may be able to shorten the treatment duration in genotype 1 HCV patients," said John McHutchison, M.D., Principal Investigator for the PROVE 1 study and Director of Gastroenterology and Hepatology Research at Duke Clinical Research Institute. "These interim results are encouraging and suggest that high sustained viral response (SVR) rates may be achieved with regimens that are 24 weeks in total duration. We look forward to 24 week follow-up data from the initial group of patients who stopped treatment at 12 weeks, and follow-up data from patients in the study who received 24 weeks of treatment."
PROVE 1 Summary
* 88% and 79% of patients receiving telaprevir achieved a rapid viral response (RVR) as measured by plasma HCV RNA <30 IU/mL and <10 IU/mL, respectively, at 4 weeks.
* Six of 9 patients in one treatment arm who completed 12 weeks of treatment, and who had achieved an RVR as defined by the study protocol (<10 IU/mL), continued to have undetectable HCV RNA 20 weeks after stopping all treatment ("SVR20").
* The treatment discontinuation rate due to adverse events through 12 weeks was 11% in telaprevir arms and 3% in the control arm. Rash, gastrointestinal events and anemia were the most common events leading to discontinuation in the telaprevir arms.
"These interim results support our approach to evaluating telaprevir-based regimens of differing durations in our Phase 2 program. The results of the 12-week duration regimen provide a level of confidence in the shorter duration approach, and we look forward to safety and antiviral data, including SVR data, from the 24-week telaprevir-based regimens," said John Alam, M.D., Executive Vice President, Medicines Development, and Chief Medical Officer of Vertex. "The information from PROVE 1 and PROVE 2 should allow us to design optimized durations and regimens for Phase 3 development."
PROVE 1 and PROVE 2 represent two of three large, ongoing clinical studies of telaprevir. In aggregrate, the three studies are designed in part to evaluate the safety and antiviral activity of different durations of telaprevir-based therapy in genotype-1 infected HCV treatment-naive and treatment-failure patients, both with and without ribavirin. Taken together, the PROVE studies are expected to provide information to optimize the treatment duration and treatment regimen for telaprevir-based therapy.
PROVE 1: Implications for Clinical Development and Registration Path
Vertex today discussed the potential implications that the new information from PROVE 1 has for future clinical development of telaprevir. Vertex stated its intention to consider evaluation of treatment regimens that would include telaprevir in combination with peg-IFN and RBV, and depending on PROVE 2 data, regimens that may exclude RBV. Vertex expects to focus on treatment durations of no more than 24 weeks. Vertex and Tibotec are planning to meet with regulatory authorities to discuss the Phase 3 design in mid-2007 and are planning to initiate Phase 3 clinical development in the fourth quarter of 2007. The registration strategy and timing of an NDA filing will be dependent on discussions with regulatory authorities.
PROVE 1 Results at EASL
Interim 12-week antiviral analysis of PROVE 1
A total of 250 patients were enrolled in PROVE 1 and received at least one dose of telaprevir or placebo in addition to Peg-interferon alfa-2a (peg-IFN) + ribavirin (RBV) in the study. A total of 175 patients received at least one dose of telaprevir in 1 of 3 arms (treatment arms B, C and D) and 75 patients received at least one dose of placebo (arm A). Treatment with telaprevir resulted in a high proportion of patients achieving a rapid viral response at 4 weeks. At the time of the interim analysis, all patients had either completed 12 weeks or discontinued from the study prior to week 12. Available 4-week and 12-week results are detailed in the following table:
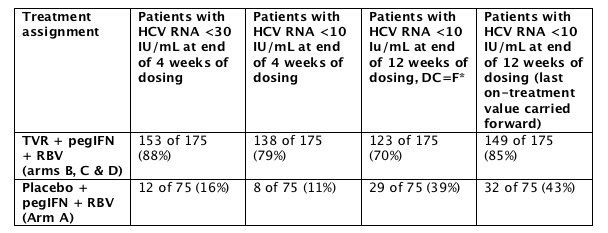
In PROVE 1, a low rate of viral breakthrough was observed. Viral breakthrough occurred in 12 patients receiving telaprevir (7%), all but one of which occurred in the first 4 weeks of treatment.
Analysis of PROVE 1 Patients who Finished All Treatment at 12 Weeks
Seventeen of 175 patients received at least one dose of telaprevir in "Arm D" of the PROVE 1 study (telaprevir + peg-IFN + RBV). According to the study protocol, patients in Arm D were eligible to stop all treatment at week 12 if they met on-treatment criteria, including the achievement of RVR (<10 IU/mL at week 4) and maintenance of this viral response (<10 IU/mL) at week 10 of treatment. Nine of 17 patients met these criteria and stopped therapy at 12 weeks, and 6 of these patients continued to have undetectable HCV RNA at week 20 of post-treatment follow-up. Of the remaining 8 patients enrolled in Arm D, 4 discontinued due to adverse events prior to week 12, and 4 did not achieve RVR.
Interim 12-Week Safety Analysis of PROVE 1
In PROVE 1, the types of adverse events that have been commonly observed with interferon and ribavirin were seen across all treatment arms. The most common adverse events, regardless of treatment assignment, were fatigue, rash, headache and nausea. Gastrointestinal disorders, rash and anemia were more common in the telaprevir arms.
In the telaprevir dosing arms, the incidence of treatment discontinuations due to adverse events through 12 weeks was 11% (19 of 175 patients), compared to 3% (2 of 75 patients) in the control arm. The difference between the two groups is due to the greater number of discontinuations due to rash, gastrointestinal disorders and anemia in the telaprevir arms compared to the control arm. The most common reason for treatment discontinuation in the telaprevir arms was rash (7 patients), and the median time to discontinuation in these patients was 64 days.
Webcast of Investor Presentation
Vertex intends to provide a live webcast of its investor presentation from Barcelona beginning at 7:30 p.m. CEST (1:30 p.m. EDT) on Saturday, April 14. The presentation may be accessed from the 'Events Calendar' on the homepage of Vertex's website at www.vrtx.com. A replay of the webcast will also be available on the Company's website until April 27, 2007. To ensure a timely connection, it is recommended that users register at least 15 minutes prior to the scheduled webcast.
About Telaprevir (VX-950)
Telaprevir (VX-950) is an investigational oral inhibitor of HCV protease, an enzyme essential for viral replication, and is one of the most advanced investigational agents in development that specifically targets HCV. Vertex is conducting a global Phase 2b clinical development program for telaprevir consisting of three large clinical trials that are expected to enroll approximately 1,000 patients with genotype-1 HCV at clinical centers in the United States, Canada and Europe. In these clinical trials, telaprevir is being dosed as 750 mg every eight hours in combination with pegylated interferon alfa-2a (Pegasys®), both with and without ribavirin (Copegus®).
Vertex retains commercial rights to telaprevir in North America. Vertex and Tibotec are collaborating to develop and commercialize telaprevir in Europe, South America, Australia, the Middle East, and other countries. Vertex is collaborating with Mitsubishi Pharma to develop and commercialize telaprevir in Japan and certain Far East countries.
About Hepatitis C
Hepatitis C is a liver disease caused by infection with hepatitis C virus (HCV), which is found in the blood of people with the disease. HCV, a serious public health concern affecting 170 million people worldwide, is spread through direct contact with the blood of an infected person. Though many people with hepatitis C may not experience symptoms, others may have symptoms such as jaundice, abdominal pain, fatigue and fever. Hepatitis C significantly increases a person's risk of developing chronic liver disease, cirrhosis, liver cancer and early death.
About Vertex
Vertex Pharmaceuticals Incorporated is a global biotechnology company committed to the discovery and development of breakthrough small molecule drugs for serious diseases. The Company's strategy is to commercialize its products both independently and in collaboration with major pharmaceutical companies. Vertex's product pipeline is focused on viral diseases, inflammation, autoimmune diseases, cancer, pain and bacterial infection. Vertex co-discovered the HIV protease inhibitor, Lexiva, with GlaxoSmithKline.
About Tibotec
Tibotec Pharmaceuticals, Ltd., based in Cork, Ireland, is a pharmaceutical research and development company. Tibotec is dedicated to the discovery and development of innovative HIV/AIDS drugs and anti-infectives for diseases of high unmet medical need. The Company's main research and development facilities are in Mechelen, Belgium with offices in Yardley, PA.
For further information on Tibotec, please visit www.tibotec.com
Safe Harbor Statement
This press release my contain forward-looking statements, including statements that (i) PROVE 1 data support potential to shorten treatment duration and increase SVR rates in patients with genotype 1 HCV infection; (ii) 12 weeks of telaprevir-based therapy enabled some patients to clear the virus; (iii) high SVR rates with telaprevir may be achieved with regimens that are no longer than 24 weeks in duration; (iv) interim results support our approach to evaluating telaprevir-based regimens of differing durations in our Phase 2 program; (v) the information from PROVE 1 and PROVE 2 will allow us to design optimized durations and regimens for Phase 3 development; (vi) the PROVE studies are expected to provide information to optimize treatment duration and treatment regimen for telaprevir-based therapy; (vii) Vertex will consider evaluation of treatment regimens that would include telaprevir in combination with peg-IFN and RBV, and depending on PROVE 2 data, regimens that may exclude RBV; (viii) Vertex expects to focus on treatment durations of no more than 24 weeks; and (ix) Vertex and Tibotec are planning to meet with regulatory authorities to discuss the Phase 3 design in mid-2007 and are planning to initiate Phase 3 clinical development in the fourth quarter of 2007. While management makes its best efforts to be accurate in making forward-looking statements, such statements are subject to risks and uncertainties that could cause the actual results of studies to vary materially. Those risks and uncertainties include, among other things, the risk that observed outcomes in clinical investigations of small numbers of patients will not be reflected in clinical trials involving larger numbers of patients, that unexpected and adverse outcomes in other ongoing clinical and nonclinical studies will occur, that the FDA or other regulatory authorities will require additional and unanticipated studies or clinical trial outcomes before granting regulatory approval, and other risks listed under Risk Factors in Vertex's Form 10-K filed with the Securities and Exchange Commission on March 1, 2007. Vertex disclaims any obligation to update the information contained in this press release as new data become available.
Vertex Contacts:
Lynne H. Brum, VP, Strategic Communications (617) 444-6614
Michael Partridge, Director, Corporate Communications, (617) 444-6108
Patricia Farrell, Director, Public Relations, (617) 444-6533
Lora Pike, Manager, Investor Relations, (617) 444-6755
Zachry Barber, Senior Media Relations Specialist, (617) 444-6470
Tibotec Contact:
Karen Manson, VP Communications and Public Affairs
Mobile: +32 479 894 799
Office: +32 15 461 019
|
| |
|
|
|
|
|