 |
|
|
| |
3-Drug HCV therapy for 12 weeks: VX950+pegIFN+RBV,
interim results from phase II PROVE 1 Trial
|
| |
| |
Reported by Jules Levin
42nd EASL, April 11-15, 2007
Sorry for the lower quality of pictures in this report but the slides were not made available to me.
At the 42nd Annual meeting for the European Association for the Study of the Liver (EASL) in Barcelona, Spain, which took place from April 11-15, 2007, the interim study results for the first oral HCV drug, an HCV protease inhibitor, captured the imagination of conference participants. Prior to the actual presentation as a Late Breaker this presentation was one of the most highly anticipated in many years. The actual presentation was the last of the session as observers waited anxiously to see the results. After the presentation it was the subject of much discussion as attendees tried to digest and remember and understand what they saw, and I am sure it will continue to be much discussed. Since the data was presented at the very end of the day at 6pm and was the last presentation attendees were tired. The results presented re-confirms the potency of VX950 and supports the treatment strategy of 12 weeks therapy with VX950 and pegIFN+ribavirin. The safety profile was characterized more than previously and showed some patients developed rash and had GI side effects. We need additional data from longer term results to better understand safety and how to use the drug but so far based on the data presented the side effects seem manageable. With longer term use often we learn how to better manage side effects of therapy. I think the results are positive based on the interim results. Of course we need to monitor the safety data. But I call these interim study results positive and encouraging.
Interim results from the phase II PROVE 1 study presented at EASL shows VX950 is a potent drug, as demonstrated by 88% of hard-to-treat genotype 1 patients who received the 3-drug telaprevir-based regimen that included Pegasys+ribavirin achieving <30 IU/mL by week 4. This achievement demonstrates the capacity of a potent oral HCV drug to achieve quick suppression of HCV and to attain high rates of Sustained Viral Responses and a 'cure' for hard-to-treat genotype 1 patients. These results are in line with prior monotherapy studies that showed VX950 median HCV RNA reduction was 4.4 log viral load reduction after several weeks of therapy. The PROVE study examined the possibility of shortening HCV therapy to 12 weeks. We already know that a shortened duration of therapy with only pegIFN + RBV has been shown in studies presented to achieve SVRs for patients. So considering the potency of adding VX950 to pegIFN+RBV achieving an SVR with a treatment strategy of 12 weeks with this triple regimen should be more easily attainable for more more patients than with pegIFN+RBV alone. The results presented at EASL suggest that for some patients 12 weeks may provide a sustained viral response. In the PROVE 1 Study, 13 of 16 patients who remained on therapy for 12 weeks achieved <30 IU/mL but only 9 met the study protocol allowing them to stop therapy after 12 weeks which was a Rapid Viral Response by week 4 <10 IU/mL. Of the total 17 patients that received drug in the 12 week therapy arm 1 patient withdrew consent and 3 discontinued due to side effects. You can read below exactly what happened in order for the patients. We need more study data to understand how to use VX950.
In the PROVE 1 Study patients taking VX950 experienced several side effects: pruritis (itching), rash, gastrointestinal, and anemia. The incidence of severe rash was about 4%, but there was no reported Stevens Johnson Syndrome. The vast majority of nausea and diarrhea was mild. The incidence of severe nausea and diarrhea was low about 2%. Anemia associated with telaprevir was grade 1 or 2. The incidence of severe declines in hemoglobin was the same for the VX950 or Peg/RBV control group. The presenter, John McHutchison, said declines in hemoglobin associated with VX950 were one half to 1 gram. Discontinuation due to adverse events was 11% in patients taking telaprevir (19/1275 patients) including 7 patients due to rash, 2 to GI disorders, 2 for anemia. This compared to a 3% rate of discontinuations due to adverse events for patients taking only Pegasys/RBV (2/75). Because the side effects of rash, anemia and gastrointestinal are common to taking ribavirin some observers speculated that there might be an interaction between VX950 and ribavirin, perhaps an increase in exposure, and so this will be examined. But I recall in early VX950 monotherapy studies rash was reported, although perhaps at a lower incidence. In sum, I think VX950 is very potent and may provide an opportunity to clear HCV for a high percentage of patients when used in combination therapy for now with peginterferon and ribavirin, but in the future perhaps along with other oral HCV drugs. It will however be years before interferon or ribavirin may be eliminated from a regimen, and perhaps it will not be possible to eliminate one or both of these drugs. The combination of VX950 plus Peg/RBV may provide up to a 90% response rate for certain patients. In the PROVE 1 Study 10% of the patients are African-American and an additional 10% are Hispanic. In this interim analysis they report 2 of the 6 patients who achieved undetectable HCV RNA 20 weeks after stopping 12 weeks therapy with VX950+Peg/RBV were African-American. It is premature to jump to conclusions but perhaps the addition of a potent oral agent such as VX950 to Peg/RBV will provide the needed antiviral potency to eradicate HCV in this hard-to-treat patient population where Sustained Viral Response rates have been about 30%. A big key to maximize VX950 success is to find a way to manage the side effects found to be apparently associated with teleprevir. If they can find a way to manage side effects to avoid or limit drug discontinuation this new therapy regimen can be even more successful. Since the results reported were 12 weeks it is too soon to understand safety and how the drug can be used in practice but after phase III we will have a better understanding of safety and efficacy and how to use VX950.
Full Data Report Filed Last Night (modified a bit)
It is late in the evening now in Barcelona at EASL as I wrap up this report because after the VX950 presentation ended at 6pm there was a symposium I attended from 6-8 on "HCV Drugs & Clinical Pharmacology". The last presentation at the conference today was the VX950 Late Breaker oral talk. This was one of the most highly anticipated presentations in recent times. This study is the first to explore 12 weeks therapy with a regimen that includes an oral HCV drug to explore the possibility of a cure with 12 weeks therapy. John McHutchison a noted researcher from Duke University presented an early look at the PROVE 1 Study, which is examining the first oral HCV drug telaprevir (VX950), an HCV protease inhibitor in combination with peginterferon plus ribavirin. Clearly VX950 demonstrates potency in this study, and oral HCV drugs will lead us into a new era of treatment. I stress that these results are interim and preliminary and we need to wait for long-term data from this study. Below is the data as presented today by McHutchison.
McHutchison Summarized: this is an interim analysis, we need the final results of the study. Significantly more patients receiving a telaprevir based regimen achieve week 4 Rapid Viral Response and week 12 undetectable status (p<0.001 at each time point) (see data below). Assuming undetectable HCV RNA at followup week 20 is maintained, the study provides proof of principle that we discussed and set out to test that a proportion of HCV genotype 1 infected patients can achieve sustained virologic response with a 12-week telaprevir-based regimen:
-- 6 of 17 subjects in ITT analysis achieved & maintained undetectable 20 weeks after stopping 12 weeks triple drug therapy (teleprevir+prg/RBV); and 6 of 9 subjects who achieved a Rapid Viral Response by week 4 were undetectable at week 12 and remained undetectable after 20 weeks following stopping therapy at week 12 (sensitive assay used: <10 & <30 IU/mL).
--low (7%) rate of 'viral breakthrough' in TVR group...sequencing for resistance ongoing.
--the most common adverse events were those that are associated with peginterferon and ribavirin.
--but by week 12 more patients in the TVR groups discontinued therapy due to adverse events (11% vs 3%) in control group), with rash as the most common reason.
--skin adverse events, gastrointestinal events and anemia occurred more commonly in the TVR groups.
--it is possible to shorten treatment duration in some treatment-naive genotype 1 HCV-infected patients with a telaprevir-based treatment regimen.
--pending final study results, improved SVR rates compared to our current standard of care may be achieved with a treatment regimen of less than 48 weeks.
--it behooves us now to focus on understanding/improving management of side effects to increase the number of patients who successfully comnplete therapy.
"Results of an Interim Analysis of a Phase 2 Study of Telaprevir (VX950) with Peginterferon alfa-2a and Ribavirin in Previously Untreated Subjects with Hepatitis C"
The PROVE 1 Study is a phase II randomized, placebo-controlled trial of previously untreated genotype 1 patients. The inclusion/exclusion criteria was standard but patients with cirrhosis were excluded. All the patients randomized received telaprevir (TVR) 750 mg every 8 hrs (q8h) or placebo, plus ribavirin 1000/1200 mg bid daily, plus Peginterferon alfa-2a (Pegasys) 180 ug subcutaneous injection weekly. The study is ongoing. A central lab is processing HCV viral loads with the Roche Taqman HCV RNA assay with a Level of Quantification (LOQ) of 30 IU/mL, and Level of Detection (LOD) of 10 IU/mL.
STUDY DESIGN
Arm A: 80 patients received Pegasys/RBV, the standard of care, and this served as the control group for 48 weeks.
Arm B: 80 patients received an initial 12 weeks of telaprevir + Peg/RBV followed by 36 weeks of Peg/RBV only for a total of 48 weeks.
Arm C: 80 patients received the 3 drugs for 12 weeks followed by 12 additional weeks of Peg/RBV.
Arm D: 20 patients receive only 12 weeks of the 3 drug regimen.
Note that subjects in arms C & D had to meet RVR (Rapid Viral Response) criterion at week 4 LOD (<10 IU/mL) and at last test before stopping therapy to stop therapy at 24 or 12 weeks. Today's presentation is an interim analysis based on all patients reaching the week 12 timepoint. Additionally they have followup data at 20 weeks now after cessation of therapy on the truncated week 12 regimen used for Arm D. For most analyses arms B, C, and D the telepravir groups are combined because the treatment for these groups was the same with the 3 drugs in the first 12 weeks. Data presented today are intent-to-treat that includes patients who received at least one dose of drugs.
Baseline Demographics
The baseline characteristics were similar between the 2 arms, control & TVR. In the TVR arm there were 10% African-Americans (n=10) and in the control arm there were 12% African-Americans. Baseline viral load was 6.6 log IU/mL (10 million IU/mL) in the TVR arm and 6.7 in the Control arm. 85% of patients had viral load >800,000 IU/mL.
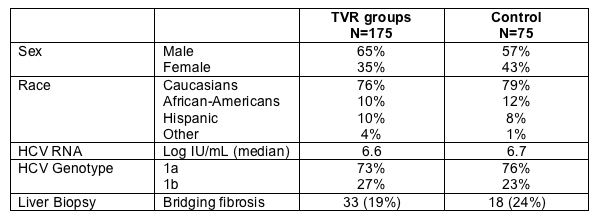
WEEK 4 HCV RNA RESULTS
Percent with Rapid Viral Response (<10 IU/mL or <30 IU/mL)
79% in the TVR triple drug arm had <10 IU/mL at week 4 compared to 11% in the Peg/RBV control arm (p<0.001). 88% in the TVR triple drug regimen achieved <30 IU/mL at week 4 compared to 16% in the Control arm (p<0.001). This was ITT analysis so patients who discontinued before week 4 were counted as non-RVR.
WEEK 12 HCV RNA RESULTS
Using the same ITT analysis at week 12, 70% in the TVR triple drug group had <10 IU/mL compared with 39% taking Peg/RBV (p<0.001). Using If the last on-treatment HCV RNA value carried forward, 85% in the TVR triple drug group had <10 IU/mL compared to 43% in the control group Peg/RBV (p<0.001).
VIRAL BREAKTHROUGHS
Definition: During the first 12 weeks of treatment either a 1-log HCV increase from nadir, or >100 Iu/mL after becoming undetectable.
14 subjects experienced viral breakthrough:
2 in the control group (3%)
12 in the TVR groups (7%)
--all but one breakthrough in the TVR groups occurred within the first month
--9 of 12 were never undetectable
sequencing for genotypic resistance in progress
GROUP D ("12 week arm")
20 patients were randomized to this arm and 3 dropped out before drug dosing so they were not counted.
17 received at least 1 dose of study drug
1 withdrew consent at week 1
16 had undetectable HCV RNA during treatment
3 discontinued for adverse events. (All these patients relapsed)
13 completed 12 weeks of TVR/peg/RBV
4 did not have an RVR at week 4 (all patients had <30 IU/mL although >10 IU/mL but according to study protocol they could not truncate their therapy at 12 weeks)
9 met the RVR criteria and stopped all treatment at week 12.
3 relapsed
6 remain undetectable 20 weeks after completion of therapy; they don't have 24 weeks yet. (2 of the 6 are African-American).
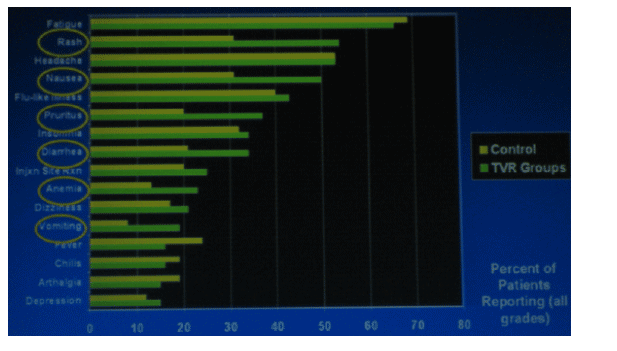
MOST COMMON ADVERSE EVENTS
There were 6 types of events that stood out as being at least 10% more common in the TVR arm than in the control arm: rash (53% vs 30%), nausea (50% vs 30%), pruritis (35% vs 20%), diarrhea (33% vs 20%), and anemia (22% vs 12).
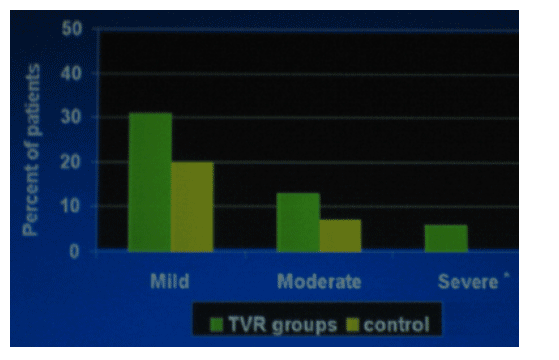
Incidence of Rash by Maximum Severity
Median time to onset of severe rash was 62 days after first dose. Severe rash which was defined by the investigators was observed in 6% of the patients taking TVR. There were no cases of mucosal involvement or Stevens Johnson Syndrome. McHutchison said anecdotally most of these rashes have resolved within a month or two.
Most Common GI Disorders
The vast majority of cases of nausea or diarrhea were mild or moderate in severity: mild nausea-- 35% TVR, 25% Control; moderate nausea-- 9% TVR, 5% Control; severe nausea-- about 4% each arm. Diarrhea: mild: 25% TVR, 17% Control, moderate: 7% TVR, 4% Control; severe: about 1% TVR.
Hemoglobin levels
Mild anemia more common in those patients receiving a TVR regimen, while the incidence of more severe anemia was similar between the TVR & control arms. More than 50% of the cases were mild or grade 2 in severity: grade 1: <11 to 9.5 (40% TVR, 29% Control; grade 2: <9.5 to 8.0: 17% TVR, 9% Control; grade 3: <8 to 6.5: about 2% in each group: grade 4: <6.5 g/dL: about 1% in each arm. McHutchison added most declines in hemoglobin on TVR were one half to 1 gram.
Proportion of Patients Discontinuing Due to Adverse Event During the First 12 Weeks
11% (n=19) of 175 patients taking TVR vs 3% (n=2) of 75 patients in Control group.
TVR
--rash (7 patients)
--gastrointestinal disorders (2)
--anemia (2)
--depression (2)
--cellulitis, dizziness, esophageal candidiasis, eye disorder, increased creatinine/BUN, mood disorder (1 each)
Control Group
GI disorder (nausea, vomiting, diarrhea); anxiety.
|
| |
|
|
|
|
|