| |
New and Emerging Treatment of Chronic Hepatitis B
|
| |
| |
Clinical Gastroenterology & Hepatology
Article in press, epub Jan 12, 2007
Emmet B. Keeffe_, Patrick Marcellin
*Division of Gastroenterology and Hepatology, Stanford University School of
Medicine, Stanford, California, and Service d'Hepatologie and INSERM CRB3,
University of Paris 7, Hopital Beaujon, Clichy, France
Corrected Proof
Conventional treatment of chronic hepatitis B with interferon alfa-2b, lamivudine, and adefovir is limited by low rates of sustained hepatitis B virus DNA suppression and hepatitis B e antigen (HBeAg) seroconversion, increasing rates of drug resistance to the oral agents, and poor tolerability of interferon. Recently several promising new antiviral agents have emerged that possess potent antiviral effects, less toxicity, and have little or no risk of drug resistance. Two new agents, entecavir and peginterferon alfa-2a, have received recent approval by regulatory authorities in the United States and several other countries for the treatment of adults with chronic hepatitis B. In large phase III clinical trials, these agents have demonstrated superior efficacy over lamivudine in both HBeAg-positive and HBeAg-negative patients. Drug resistance occurs at a low rate in lamivudine-refractory patients treated with entecavir or is, to date, nonexistent in nucleoside-naive patients treated with entecavir and all patients receiving peginterferon. In addition, several novel agents in clinical development, such as emtricitabine, clevudine, telbivudine (has been approved), valtorcitabine, and tenofovir, have shown promising clinical profiles in patients with chronic hepatitis B. This review summarizes the recent clinical studies of these new agents and discusses the implications of these data for the management of chronic hepatitis B.
Chronic hepatitis B virus (HBV) infection is an important cause of liver disease worldwide, affecting approximately 400 million individuals who are at risk for the sequelae of cirrhosis, hepatic decompensation, and hepatocellular carcinoma (HCC).1, 2 The goal of treatment of chronic hepatitis B (CHB) is to achieve sustained suppression of HBV DNA replication and improvement of liver necroinflammation with normalization of serum alanine aminotransferase (ALT) levels before liver fibrosis progresses to an advanced and irreversible stage. Before 2005, conventional treatments for individuals with chronic HBV infection included parenterally administered interferon (IFN) alfa-2b, the oral nucleoside analog lamivudine, and the oral nucleotide analog adefovir dipivoxil. Adefovir was a significant advance,3, 4 as compared with lamivudine, in terms of decreasing the selection of antiviral-resistant HBV mutants from 60% to 70% with lamivudine to 15% and 29% with adefovir after 4-5 years of therapy.5, 6
Data from long-term cohort studies of individuals chronically infected with HBV show that decrease of viral replication to low levels, with reduction or prevention of hepatic injury, is fundamental to prevention of disease progression and the prolongation of survival.7, 8 Antiviral therapy with IFN or lamivudine has been shown to halt disease progression, reverse hepatic fibrosis, reduce the complications of cirrhosis, and prolong survival.9, 10, 11, 12, 13, 14 More recent evidence from clinical and community-based studies suggests that the serum HBV DNA level is a predictor of the risk for cirrhosis and HCC, independent of hepatitis B e antigen (HBeAg) status and serum ALT levels.15, 16, 17, 18, 19 These data support the importance of achieving sustained suppression of HBV viral replication to slow or prevent disease progression. Unfortunately, sustained suppression of HBV DNA is often not achieved with nucleoside antiviral therapies such as lamivudine and adefovir because of the emergence of drug resistance and/or suboptimal suppression of viral replication. In addition, the use of IFN is limited by inconvenience of injection, poor tolerability, and high cost.
Recently, several drugs with increased potency, improved tolerability, and little or no drug resistance have been evaluated for the treatment of patients with CHB. This review will focus on clinical data regarding the recently licensed nucleoside analog entecavir and peginterferon alfa-2a. Data on other promising therapeutic approaches in late-stage clinical development will also be reviewed.
Abbreviations used in this paper: ALT, alanine aminotransferase; anti-HBe, antibody to HBeAg; bDNA, branched DNA; CHB, chronic hepatitis B; HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HIV, human immunodeficiency virus; INF, interferon; PCR, polymerase chain reaction; ULN, upper limits of normal.
Recently Licensed Therapies for Chronic Hepatitis B
Entecavir
Entecavir, a potent deoxyguanosine nucleoside analog with antiviral activity specific for hepadnaviruses, is the newest oral antiviral agent against HBV.20, 21 Entecavir triphosphate, the active intracellular form of entecavir, inhibits HBV DNA polymerase more effectively than lamivudine or adefovir. In vitro, entecavir triphosphate demonstrates a higher binding affinity for HBV DNA polymerase than the natural guanosine triphosphate substrate and effectively inhibits HBV DNA replication at 3 stages in the replication pathway. In addition to inhibiting the reverse transcription of the negative-strand HBV DNA from the pregenomic messenger RNA and the synthesis of the positive-strand HBV DNA, entecavir effectively suppresses the priming of HBV DNA polymerase, a step involving the covalent linkage with guanosine triphosphate. In in vitro cell culture assays with a human hepG2.2.15 hepatoma cell line, entecavir was 30-fold more potent than lamivudine in suppressing HBV viral replication, with a median effective dose of 0.004 μmol/L compared with 0.116 μmol/L for lamivudine.20 Entecavir has also been shown to be effective against lamivudine-resistant HBV at concentrations 20-fold to 30-fold higher than those required to inhibit wild-type HBV.22
The potential therapeutic benefits of entecavir have been demonstrated in woodchuck and duck models of HBV23, 24, 25 and was confirmed in phase I/II preclinical studies.26, 27, 28 In these initial clinical studies, entecavir demonstrated potent dose-dependent antiviral activity in patients with HBeAg-positive and HBeAg-negative as well as lamivudine-refractory CHB. In patients with HBeAg-positive and HBeAg-negative CHB, entecavir 0.1 and 0.5 mg/day for 24 weeks was superior to lamivudine 100 mg/day in reducing viral load (-0.97 log10 for entecavir 0.1 mg/day and -1.28 log10 for entecavir 0.5 mg/day vs lamivudine; P < .0001 for both comparisons).27 Subgroup analysis based on pretreatment ALT levels (categorized as <1.25 times the upper limits of normal (ULN), between 1.25-2.5 times ULN, or ≥2.5 times the ULN) revealed that median log10 decline in HBV DNA by polymerase chain reaction (PCR) was similar among all subgroups. These data suggest that entecavir is effective in reducing HBV DNA, regardless of baseline ALT levels. In a randomized, dose-ranging, phase II study involving patients with lamivudine-resistant CHB who were viremic despite lamivudine treatment for up to 24 weeks or had documented lamivudine resistance substitutions, entecavir was superior to lamivudine in suppressing HBV DNA to undetectable levels (by bDNA assay) (79% for 1.0 mg entecavir, 51% for 0.5 mg entecavir, 13% for lamivudine; P < .0001).28 After 48 weeks, mean reductions in HBV DNA levels were 2.85, 4.46, and 5.06 log10 copies/mL in patients who received entecavir 0.1, 0.5, and 1.0 mg, respectively, all significantly greater than the decrease of 1.37 log10 copies/mL afforded by lamivudine. In this study, higher proportions of patients achieved ALT normalization while receiving entecavir 0.1, 0.5, and 1.0 mg (47%, 59%, and 68%, respectively) compared with lamivudine (6%). Virologic rebound caused by resistance was observed in 1 patient in the 0.5 mg entecavir group.28 (3 years resistance data has been publicly presented, AASLD 2006, and no entecavir resistance has been reported in treatment-naives; 4 year update expected soon).
These preliminary findings have recently been confirmed in large phase III studies comparing entecavir and lamivudine in patients with CHB who were nucleoside-naive or were lamivudine-refractory. In these studies, entecavir was superior to lamivudine in improving liver histology, normalizing ALT levels, and reducing HBV DNA levels in nucleoside-naive HBeAg-positive and HBeAg-negative patients and lamivudine-refractory HBeAg-positive patients.29, 30, 31 Chang et al28 conducted a multinational, double-blind, phase III study comparing entecavir 0.5 mg once daily with lamivudine 100 mg once daily, administered for ≥52 weeks to 715 nucleoside-naive HBeAg-positive patients. The primary efficacy end point was histologic improvement (decrease by ≥2 points in the Knodell necroinflammatory score, without worsening of fibrosis) at week 48. Secondary end points included a reduction in the serum HBV DNA level, HBeAg loss and seroconversion, and normalization of the ALT level. At week 48, histologic improvement occurred in a higher proportion of patients in the entecavir group than in the lamivudine group (72% vs 62%, respectively; P = .009) (Table 1).29 More patients in the entecavir group than in the lamivudine group also achieved undetectable serum HBV DNA levels by the branched DNA (bDNA) assay (67% vs 36%, respectively; P < .001), normalization of ALT levels (68% vs 60%, respectively; P = .02), and greater mean reduction in serum HBV DNA from baseline (6.9 vs 5.4 log10 copies/mL, respectively; P < .001) (Table 2).29 The rate of HBeAg loss was similar for both treatment groups (22% entecavir vs 20% lamivudine), as was the rate of HBeAg seroconversion (21% entecavir vs 18% lamivudine). No evidence of resistance to entecavir was detected at week 48 of treatment. Overall, the data from this study support a significant clinical advantage of entecavir over lamivudine in the treatment of patients with HBeAg-positive CHB, because entecavir appears both more potent and associated with a lower rate of resistance.
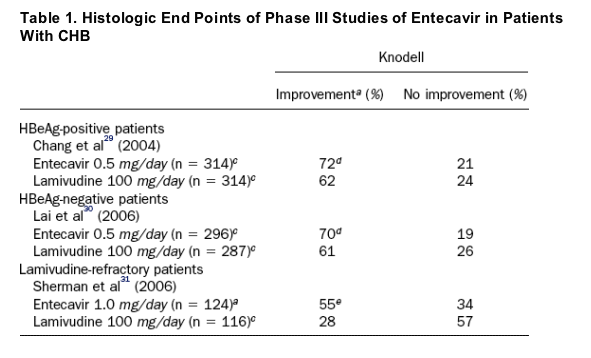
aDecrease in Knodell necroinflammatory score of 2 points from baseline with no worsening of the Knodell fibrosis score.
bFor Ishak fibrosis score, improvement was defined as a decrease of 1 point from baseline and worsening as an increase of 1 point from baseline.
cPatients with evaluable baseline histology (baseline Knodell necroinflammatory score 2).
dP .05.
eP .0001.
fP .0019.

In another study, the antiviral effects of entecavir were compared with those of lamivudine in 648 nucleoside-naive patients with HBeAg-negative CHB.30 Patients were randomized to receive entecavir 0.5 mg/day or lamivudine 100 mg/day for up to 96 weeks. The primary end point was the percentage of patients at week 48 with a reduction in Knodell necroinflammatory score of ≥2 points and no worsening of fibrosis (increase in Knodell fibrosis score of ≥1 point). As in the previously described study, the rates of histologic improvement, virologic response, and normalization of ALT levels were significantly higher at 48 weeks in patients who received entecavir than in those treated with lamivudine. At week 48, a higher proportion of entecavir-treated than lamivudine-treated patients who had adequate baseline liver biopsy specimens had histologic improvement (70% vs 61%, respectively; P = .01) (Table 1).30 In addition, more patients in the entecavir group than in the lamivudine group had undetectable serum HBV DNA levels by a PCR assay, normalization of ALT levels, a greater mean reduction in serum HBV DNA levels from baseline (Table 2). There was no evidence of resistance to entecavir. Data from this study confirm preliminary data from earlier phase II studies that entecavir is superior to lamivudine in the treatment of patients with HBeAg-negative CHB.
A third multinational phase III study compared entecavir and lamivudine in lamivudine-resistant patients with HBeAg-positive CHB (Table 1).31 In this study, 286 HBeAg-positive patients who were refractory to lamivudine therapy (persistent viremia or documented YMDD mutations while receiving lamivudine) were randomized to switch to entecavir 1 mg daily or continue lamivudine 100 mg daily for a minimum of 52 weeks. Two co-primary end points were assessed at 48 weeks, histologic improvement and a composite end point of HBV bDNA <0.7 mEq/mL and ALT <1.25 times ULN. As with previous phase III studies involving nucleoside-naive patients, the principal findings of this study favored entecavir over lamivudine. Histologic improvement occurred in 55% of entecavir-treated compared with 28% of lamivudine-treated patients (P < .0001) (Table 1).31 More patients on entecavir than on lamivudine also achieved the composite end point, 55% (77/141) compared with 4% (6/145), respectively (P < .0001). Mean change from baseline in HBV DNA was -5.11 log10 copies/mL for entecavir-treated patients and -0.48 log10copies/mL for lamivudine-treated patients (P < .0001). Virologic rebound because of entecavir-resistance substitutions occurred in 2 of 141 entecavir-treated patients, and genotypic evidence of resistance was detected in 10 patients. The safety profile of entecavir was comparable to lamivudine, with fewer ALT flares on treatment.
Long-term efficacy data indicate that the virologic (HBV DNA levels of <0.7 mEq/mL by bDNA assay, and HBeAg loss for HBeAg-positive patients; HBV DNA levels of <0.7 mEq/mL by bDNA assay, and ALT ≥1.25 times ULN for HBeAg-negative patients) and biochemical responses achieved with entecavir at week 48 in HBeAg-positive and -negative nucleoside-naive patients are sustained with continued treatment until week 96.32, 33, 34 During the second year of treatment, the proportion of entecavir-treated HBeAg-positive patients with undetectable HBV DNA (DNA levels of <300 copies/mL) increased from 64% to 81%, whereas the proportion of patients with undetectable HBV DNA in the lamivudine group remained unchanged at 30%. At 96 weeks, a higher proportion of entecavir-treated patients than lamivudine-treated patients sustained HBV DNA levels of <300 copies/mL (80% entecavir-treated patients vs 39% lamivudine-treated patients; P < .0001).32 The cumulative rate of HBeAg seroconversion (31% entecavir-treated patients vs 26% lamivudine-treated patients) and ALT normalization, defined as ≦1 _ ULN (87% entecavir-treated patients vs 79% lamivudine-treated patients) during the 2 years of therapy was comparable between the entecavir and lamivudine groups, respectively. Similar findings were reported in a second study that compared long-term entecavir and lamivudine therapy in HBeAg-negative nucleoside-naive patients.33 In this study, HBeAg-negative patients who achieved a response (HBV DNA levels of <0.7 mEq/mL by bDNA assay, and ALT <1.25 times ULN) at week 48 and discontinued therapy were followed for an additional 24 weeks. More patients treated with entecavir than lamivudine maintained HBV DNA levels of <300 copies/mL and ALT levels of ≦1 _ ULN during 24 weeks of treatment follow-up period. For those patients who experienced viral rebound off treatment, retreatment with entecavir for 12 weeks resulted in a profound viral suppression. At week 48, 90% of patients who restarted therapy with entecavir achieved HBV DNA levels of <300 copies/mL. In a third study of long-term therapy with entecavir in patients with lamivudine-refractory CHB, prolonged treatment with entecavir was also more effective than lamivudine in maintaining HBV DNA levels of <300 copies/mL (30% vs 1%, respectively; P < .0001), ALT normalization defined as ≦1 _ ULN (85% vs 29%, respectively; P < .0001), and HBeAg seroconversion rates (16% vs 4%, respectively; P = .001).34
Overall, these studies demonstrate that after 48 weeks of treatment, entecavir is superior to lamivudine in its ability to reduce HBV DNA to undetectable levels, normalize ALT, and improve liver histology in nucleoside-naive HBeAg-positive and -negative patients and lamivudine-refractory HBeAg-positive patients. The virologic and biochemical efficacy of entecavir is maintained with prolonged treatment for up to 96 weeks. In addition, restarting entecavir effectively suppresses viral replication in nucleoside-naive HBeAg-negative patients who had achieved a response and subsequently experienced viral rebound off therapy. Safety analysis from phase III studies shows that entecavir and lamivudine possess similar safety profiles. The resistance data for entecavir show no evidence of resistance in nucleoside-naive hepatitis B surface antigen (HBsAg)-positive and -negative patients treated for up to 2 years and a low rate of resistance (1% at 1 years and 9% at 2 years) in lamivudine-refractory patients.35 (data after 3-years entecavir therapy showed no resistance, 2006 AASLD; 4-year data is expected soon).
In March 2005 entecavir received approval from the U.S. Food and Drug Administration for the treatment of adults with CHB. Entecavir dosages of 0.5 mg/day are recommended for treatment-naive patients, whereas a higher dosage of 1 mg/day is recommended in adult patients and adolescents (≥16 years of age) with a history of HBV viremia who are receiving lamivudine or have known lamivudine resistance mutations.
Peginterferon-alpha
Two forms of peginterferon alpha, alfa-2a and alfa-2b, created by attaching a polyethylene glycol molecule to IFN alfa-2a or alfa-2b, differ with respect to receptor binding and pharmacologic properties. The peginterferons exhibit dual immunomodulatory and antiviral mode of action similar to the conventional IFNs but possess an improved pharmacokinetic profile. The longer serum half-life of the peginterferons allows for convenient once weekly dosing and better maintenance of effective IFN concentrations throughout the dosing interval, as compared with the conventional IFNs.36 In clinical trials of patients with chronic hepatitis C, peginterferons yielded superior clinical outcomes to conventional IFNs.37, 38 These findings prompted the evaluation of peginterferon in the treatment of patients with CHB.
Results of several large randomized studies of peginterferon in patients with CHB have recently been published (Table 3).39, 40, 41, 42 Janssen et al39 compared the safety and efficacy of peginterferon alfa-2b alone and in combination with lamivudine in patients with HBeAg-positive CHB. A total of 307 patients were randomized to receive peginterferon alfa-2b 100 _g/wk for 32 weeks, followed by 50 _g/wk for 20 weeks with placebo, alone or in combination with lamivudine 100 mg/day for 52 weeks. Patients were followed for an additional 26 weeks after treatment. At 52 weeks, more patients in the combination therapy group than in the monotherapy groups had HBV DNA levels of <200,000 copies/mL and normalization of ALT levels (Table 3). Similarly, a higher proportion of patients who received combination therapy than who received monotherapy lost HBeAg (44% vs 29%; P = .01).39 However, loss of HBeAg was comparable between the 2 groups at the end of follow-up (35% in the combination therapy group vs 36% in the monotherapy group; P = .91) as a result of a higher rate of relapse in the combination therapy group. Thus, the final results showed that combination therapy with peginterferon alfa-2b plus lamivudine was not superior to peginterferon alfa-2b monotherapy.
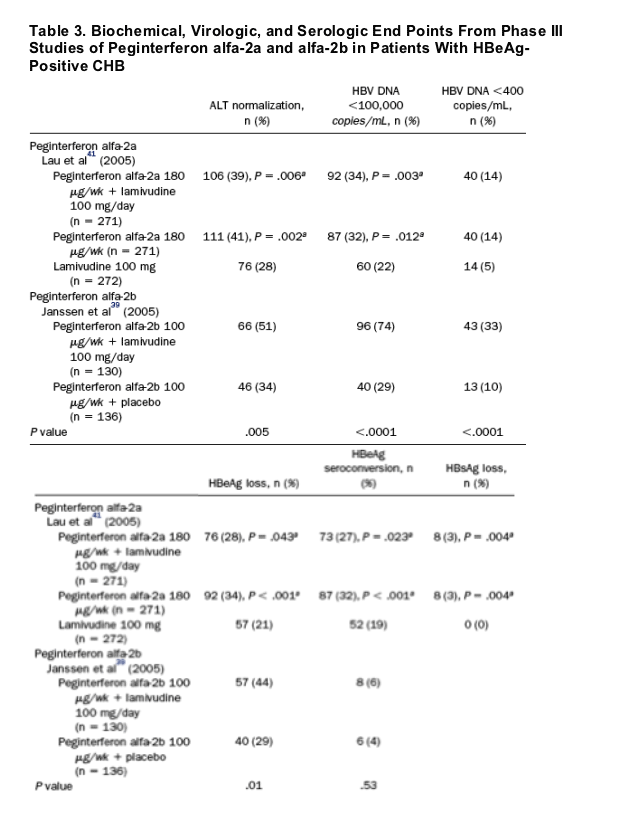
A second study by Chan et al40 compared the combination of peginterferon alfa-2b with lamivudine against lamivudine monotherapy in treatment-naive patients with HBeAg-positive CHB. In this study 100 patients were randomized to receive peginterferon alfa-2b 1.5 _g/kg body weight per week for 32 weeks plus lamivudine 100 mg/day for 52 weeks, or the same regimen of lamivudine alone for 52 weeks. The virologic response (defined as HBeAg seroconversion, detection of antibody to HBeAg, and HBV DNA levels <5 _ 105 copies/mL) and biochemical response (defined as normalization of ALT levels) were assessed at end of treatment and at 24 weeks after the end of treatment. At the end of treatment, a higher rate of virologic response was reported after combination therapy than after lamivudine monotherapy (60% vs 28%, respectively; P = .001), as was a higher median reduction in HBV DNA levels (3.91 vs 2.83 log10 copies/mL, respectively).43 Patients who received combination therapy were also less likely to develop lamivudine resistance than were those who received lamivudine monotherapy (21% vs 40%, respectively; P = .045). The percentage of patients achieving normalization of ALT levels and histologic improvement was similar between the 2 treatment groups.
Peginterferon alfa-2a alone and in combination with lamivudine has also been assessed in patients with HBeAg-positive and -negative CHB. Lau et al41 reported the results of a phase III study evaluating peginterferon alfa-2a in 814 patients with HBeAg-positive CHB. Patients were randomized to receive peginterferon alfa-2a 180 _g/wk alone, peginterferon alfa-2a 180 _g/wk plus lamivudine 100 mg/d, or the same regimen of lamivudine alone for 48 weeks and were followed for an additional 24 weeks after the end of treatment. The primary efficacy end point of the study defined as HBeAg seroconversion, detection of antibody to HBeAg, and HBV DNA levels of <5 _ 105 copies/mL by using Cobas Amplicor (Roche Molecular Diagnostics, Basel, Switzerland). HBV test was assessed after 24 weeks of treatment-free follow-up. The majority of patients in this study were Asian (87%), and most (87%-88%) were infected with HBV genotype B or C. After 24 weeks of follow-up, HBeAg seroconversion was reported in significantly more patients who received peginterferon alfa-2a as monotherapy (32%) or in combination with lamivudine (27%) than in those who received lamivudine monotherapy (19%; P < .001 compared with peginterferon alfa-2a monotherapy, P = .02 compared with combination therapy).41 Similarly, HBV DNA levels of <1.0 _ 105 copies/mL were reported in a significantly greater percentage of patients who received peginterferon alfa-2a as monotherapy (32%) or in combination with lamivudine (34%) than in patients who received lamivudine monotherapy (22%; P = .01 compared with peginterferon alfa-2a monotherapy, P = .003 compared with combination therapy). Sixteen patients receiving peginterferon alfa-2a alone or in combination underwent HBsAg seroconversion, compared with 0 patients receiving lamivudine alone (P = .001). Serious adverse events occurred in 4%, 6%, and 2% of patients receiving peginterferon alfa-2a monotherapy, combination therapy, and lamivudine monotherapy, respectively. Two patients receiving lamivudine monotherapy experienced irreversible liver failure after the cessation of treatment; one underwent liver transplantation, and the other died.
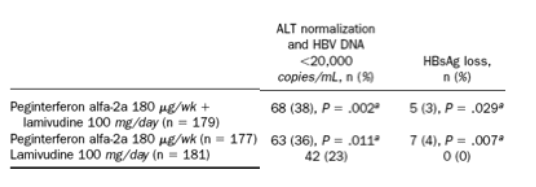
A second study assessed the efficacy and safety of peginterferon alfa-2a therapy in patients with HBeAg-negative CHB.42 A total of 537 patients were randomly assigned to 1 of 3 treatment groups: peginterferon alfa-2a monotherapy 180 _g/wk, peginterferon alfa-2a 180 _g/wk plus lamivudine 100 mg/day, or the same regimen of lamivudine alone for 48 weeks. The 2 co-primary end points of the study, normalization of ALT and suppression of HBV DNA levels of <2.0 _ 104 copies/mL, were assessed after 24 weeks of follow-up. At the end of follow-up, normalization of ALT levels and HBV DNA levels of <2.0 _ 104copies/mL were achieved by a greater percentage of patients in the peginterferon alfa-2a monotherapy group (59% and 43%, respectively) and the combination therapy group (60% and 44%, respectively) than in the lamivudine monotherapy group (44%, P = .004 and P =.003, respectively; and 29%, P = .007 and P = .003, respectively) (Figure 1).42 A higher proportion of patients in the peginterferon alfa-2a monotherapy group and the combination therapy group than in the lamivudine monotherapy group also achieved HBV DNA levels of <400 copies/mL (19% vs 20% vs 7%, respectively; P < .001 for both comparisons to lamivudine alone) and HBsAg loss (12 patients in peginterferon groups and 0 in lamivudine group) (Table 4).42 Peginterferon alfa-2a monotherapy and combination therapy also resulted in a greater reduction in HBV DNA levels than did lamivudine monotherapy. Moreover, rates of sustained response, defined as sustained HBV DNA suppression, were higher with peginterferon alfa-2a monotherapy and combination therapy with lamivudine than with lamivudine monotherapy (19% vs 20% vs 7%; P < .001 for both comparisons to lamivudine).42 Adverse events, including pyrexia, fatigue, myalgias, and headache, were less frequent with lamivudine monotherapy than with peginterferon alfa-2a monotherapy or combination therapy.
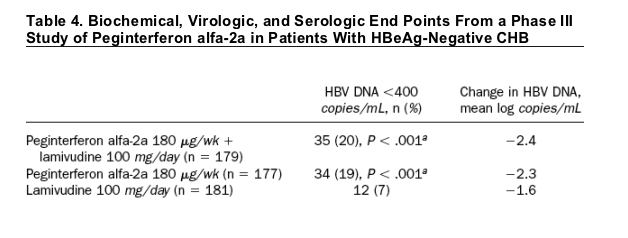
Data from long-term follow-up studies suggest that the virologic response to peginterferon alfa-2a is sustained after treatment in patients with HBeAg-positive CHB and HBeAg-negative CHB.44, 45 Lau et al44 assessed the durability of response to peginterferon alfa-2a monotherapy in cohort of Asian patients who underwent HBeAg seroconversion 6 months after treatment. Of the 58 patients who seroconverted at 6 months, 48 (83%) maintained seroconversion at 12 months after treatment. An additional 3 (5%) patients remained HBeAg-negative but lost antibody to HBeAg (anti-HBe) by the 1-year follow-up. In this study, 15% of Asian patients who did not HBeAg seroconvert at 6 months after treatment did undergo seroconversion by 12 months.44 In this analysis sustained HBeAg seroconversion was associated with higher baseline ALT and lower baseline HBV DNA levels. A second study assessed factors associated with sustained response to peginterferon in 177 patients with HBeAg-negative CHB.45 At 48 weeks after treatment, 49 (55%) of the 89 patients who had a virologic response had sustained HBV DNA levels of <105 copies/mL during the 48-week follow-up period, including 22 (25%) with HBV DNA levels between 2 _ 104 and 2 _ 105 copies/mL and 27 (30%) with HBV DNA levels of <20,000 copies/mL.45 Among the patients achieving an ALT response, 72% maintained ALT levels at <1.5 _ ULN at 6-12 months after treatment. At the end of the initial 6 months of therapy, 7 patients had achieved HBsAg loss or seroconversion, and 1 additional patient with a sustained virologic response had achieved HBsAg loss or seroconversion between 6-12 months.45
Overall findings from these studies demonstrate that peginterferon offers superior efficacy to lamivudine, resulting in a greater incidence of HBeAg seroconversion, HBV DNA suppression, and HBsAg seroconversion in patients with HBeAg-positive and -negative CHB, particularly in those with lower baseline HBV DNA and high baseline ALT levels. Patients with HBeAg-negative CHB had significantly higher rates of response, sustained for 24 weeks after the cessation of therapy, with peginterferon alfa-2a than with lamivudine. The addition of lamivudine to peginterferon alfa-2a did not improve post-therapy response rates; however, more studies are needed to assess the safety and efficacy of the combination of peginterferon plus other oral antiviral agents. Larger controlled clinical trials are needed to assess the value of this type of combination. Peginterferon therapy was not associated with the development of drug resistance in any of the trials described above. Tolerance of peginterferon therapy in these trials appeared to be as good as or better than tolerance seen in the hepatitis C trials, on the basis of the data reported on adverse events. Peginterferon alfa-2a 180 _g/wk subcutaneously is approved in the United States and Europe for the management of HBeAg-positive or -negative CHB in adults with compensated liver disease and evidence of viral replication, elevated ALT levels, and histologically verified liver inflammation and/or fibrosis. Additional studies are required to determine the long-term effects of peginterferon on the development of cirrhosis and HCC and its use in combination with other antiviral agents.
Agents in Clinical Development
Ongoing phase II/III trials are investigating several new types of drugs for the management of chronic HBV infection. These include emtricitabine, clevudine, telbivudin (approved in Fall 2006 after this article was written), valtorcitabine, and tenofovir (Table 5). All appear to be under development for possible regulatory approval for the treatment of CHB, and some are currently available for the treatment of human immunodeficiency virus (HIV) infection.

Emtricitabine (FTC)
Emtricitabine, a nucleoside analogue of 2_-deoxycytidine 5_-triphosphate that is structurally similar to lamivudine, exhibits activity against both HBV and HIV. Currently, emtricitabine is approved in the United States for use in combination with other antiretroviral agents for the management of HIV infection in adults aged ≥18 years.
Two randomized phase II studies have evaluated emtricitabine in patients with HBeAg-positive or -negative CHB.46, 47 In a double-blind, parallel-group study, Gish et al46 evaluated the long-term safety and antiviral activity of emtricitabine in HBeAg-positive nucleoside-naive patients. Patients were randomized to receive emtricitabine 25, 100, or 200 mg/day for 48 weeks. At the end of treatment, patients were allowed to continue receiving emtricitabine 200 mg on an open-label basis for an additional 48 weeks. Study end points included reductions in serum HBV DNA and ALT levels and rates of HBeAg seroconversion. After 2 years, 53% of the patients had serum HBV DNA levels of ≦4.7 _ 103 copies/mL, 33% seroconverted to anti-HBe, and 85% had achieved normalization of ALT levels.46 The incidence of L526M ± M550V/I-associated resistance was 18% for patients receiving emtricitabine 200 mg for the full 2 years, a figure about half of that seen with lamivudine, which suggests that resistance rates are lower with emtricitabine.
A second study compared the safety and efficacy of emtricitabine with placebo in nucleoside/nucleotide-naive patients with HBV.47 Patients were randomized to receive emtricitabine 200 mg/day or placebo once daily for 48 weeks and underwent a pretreatment and end-of-treatment liver biopsy. Histologic improvement was defined as a 2-point reduction in Knodell necroinflammatory score with no worsening in fibrosis. At the end of treatment, more patients receiving emtricitabine than placebo had improved liver histologic findings (62% vs 25%, respectively; P < .001), serum HBV DNA levels <400 copies/mL (54% vs 2%, respectively; P < .001), and normalization of ALT levels (65% vs 25%, respectively; P < .001).47 The rate of seroconversion to anti-HBe (12%) and HBeAg loss were similar between arms. At week 48, resistance mutations were detected in 20 (13%) of 159 patients in the emtricitabine group. The safety profile of emtricitabine during treatment was similar to that of placebo. Post-treatment exacerbation of HBV infection developed in 23% of emtricitabine-treated patients.47
Emtricitabine was well-tolerated and demonstrated a potent antiviral response for up to 2 years in patients with CHB; on the basis of these data, emtricitabine 200 mg/day was chosen as the optimal dose for future studies of patients with CHB. Although emtricitabine is effective at inhibiting HBV replication, drug resistance and cross-resistance with lamivudine might limit its role as a single agent in the management of CHB. Ongoing studies are examining emtricitabine in combination with other anti-HBV agents such as adefovir, clevudine, and tenofovir.
More recently, Lim et al48 evaluated the antiviral effects of emtricitabine in combination with clevudine in patients with CHB. In this study, 163 patients who had completed the phase 3 study of emtricitabine were randomized to receive emtricitabine 200 mg/day plus clevudine 10 mg/day or emtricitabine 200 mg/day plus placebo for 24 weeks with 24 weeks of follow-up. Serum HBV DNA levels were assessed after 24 weeks by using the Digene HBV Hybrid Capture II (Digene Corporation, Gaithersburg, MD) assay. After 24 weeks of treatment, more patients receiving combination therapy than monotherapy achieved HBV DNA levels of <4,700 copies/ml (74% vs 65%, respectively; P = .114), undetectable viremia (40% vs 23%, respectively; P ≥ .025), and normalization of ALT levels (63% vs 42%, respectively; P ≥ 0.025).48 The safety profile was similar between arms during treatment, with less post-treatment exacerbation of hepatitis B in the combination arm.
Clevudine
Clevudine is a pyrimidine nucleoside analog that has demonstrated potent activity against HBV in the woodchuck HBV model49 and more recently in clinical trials of patients with CHB.50, 51 A multicenter dose-escalation study has shown that clevudine 10, 50, 100, and 200 mg/day for 28 days causes potent and durable post-treatment viral suppression in patients with CHB infection.50 Patients in this study had HBV DNA levels of ≥3 _ 106 copies/mL, had not undergone nucleoside treatment, and were without HIV or HCV coinfection. Thirty-two patients were enrolled (5, 10, 10, and 7 patients in the 10, 50, 100, and 200 mg/day groups, respectively), 81% were male, 81% were Asian, and 88% were HBeAg-positive at baseline. Median pretreatment serum HBV DNA levels ranged from 7.3-8.8 log10 copies/mL. After 28 days, the median HBV DNA change from baseline was -2.5, -2.7, -3.0, and -2.6 log10 copies/mL in the 10-, 50-, 100-, and 200-mg cohorts, respectively, and 6 months after dosing, median changes from baseline were -1.2, -1.4, -2.7, and -1.7 log10 copies/mL, respectively.50 Six of 27 patients lost HBeAg, and 3 of 27 patients seroconverted to anti-HBe. Clevudine was well-tolerated, with no dose-limiting toxicities. A transient increase in ALT levels of up to 7.8 times (increase of 20-186 IU/L) was observed in 6 patients in the 100-mg cohort, without signs of liver failure. These increases were associated with improved viral suppression. The pharmacokinetic profile of clevudine was proportional to the dose. These results demonstrate the tolerability and potent activity of clevudine in HBV-infected patients and support further clinical study.
Lee et al51 recently assessed the safety, tolerability, and antiviral response to clevudine in 98 patients with HBeAg-positive CHB. In this study, clevudine showed potent antiviral activity during therapy and induced a sustained post-treatment antiviral effect for 6 months after a 12-week treatment period that was associated with a sustained normalization of ALT levels.51 Patients were randomized to placebo, clevudine (30 mg), and clevudine (50 mg) once daily for 12 weeks and followed up for 24 weeks off therapy. At week 12, median serum HBV DNA reductions from baseline were 0.20, 4.49, and 4.45 log10 copies/mL in the placebo, clevudine 30 mg, and clevudine 50 mg groups, respectively (P < .0001).51 Post-treatment antiviral activities were sustained at weeks 12 and 24 off therapy. Marked reductions in median serum ALT levels occurred during clevudine treatment and were maintained below ULN throughout the 24 weeks off therapy in the 2 clevudine-treated groups. The incidences of adverse events and treatment-emergent grade 3 or 4 laboratory abnormalities were similar for the 3 groups.
Telbivudine and Valtorcitabine
Telbivudine is an HBV-specific L-nucleoside analog of thymidine. Telbivudine specifically targets the viral DNA polymerase enzyme that is responsible for HBV replication and demonstrates no activity against other viruses that cause human disease. Despite their close structural relationship, telbivudine, lamivudine, and valtorcitabine have unexpected differences in their modes of action.52 Valtorcitabine and lamivudine both preferentially inhibit first-strand (RNA-dependent) DNA synthesis, whereas telbivudine preferentially inhibits HBV second-strand (DNA-dependent) DNA synthesis.53
Several studies have examined the efficacy and safety of telbivudine54, 55, 56 and valtorcitabine57 in patients with CHB. In phase I/II studies, telbivudine demonstrated marked dose-related antiviral activity at dosages of ≥400 mg/day.54 A randomized, double-blind, multicenter trial evaluated the efficacy and safety of telbivudine 400 or 600 mg/day and telbivudine 400 or 600 mg/day plus lamivudine 100 mg/day (Comb400 and Comb600) compared with lamivudine 100 mg/day in 104 HBeAg-positive adults with compensated CHB.55 At week 52, patients treated with telbivudine monotherapy exhibited a greater mean reduction in HBV DNA levels (6.01 vs 4.57 log10 copies/mL; P < .05), clearance of PCR-detectable HBV DNA (61% vs 32%; P < .05), and normalization of ALT levels (86% vs 63%; P < .05) compared with those who received lamivudine monotherapy, with proportionally greater incidence of HBeAg seroconversion (31% vs 22%) and less viral breakthrough (4.5% vs 15.8%; P = NS for both). Combination treatment was not better than telbivudine monotherapy. All treatments were well-tolerated.
These findings were confirmed by data from a large, randomized, phase III trial (GLOBE study) of telbivudine and lamivudine in 1367 patients with HBeAg-positive and -negative CHB.56 A total of 1367 HBeAg-positive and HBeAg-negative patients were randomized to treatment with telbivudine 600 mg/day or lamivudine 100 mg/day. At week 52, telbivudine was more effective than lamivudine in achieving all virologic efficacy end points in both HBeAg-positive and -negative patients (P < .01).56 In the HBeAg-positive intent-to treat population, telbivudine was superior to lamivudine for the primary efficacy end point of therapeutic response (HBV DNA levels of <5 log10 plus HBeAg loss or ALT normalization), both at weeks 52 and 76. In the HBeAg-negative intent-to-treat population, response (HBV DNA levels of <5 log10 plus HBeAg loss or ALT normalization) at weeks 52 and 76 was similar between the treatment groups. Telbivudine demonstrated a safety profile comparable to that of lamivudine and was associated with lower rates of primary treatment failure, lower rates of development of resistance, and fewer and less severe instances of ALT flares than lamivudine.56
A recent multivariate subanalysis of the GLOBE study found that telbivudine provided better virologic responses than did lamivudine across nearly all patient subgroups.58 Greater HBV DNA suppression was observed with telbivudine in HBeAg-positive patients with baseline ALT levels of >2.5 _ ULN than in patients with baseline ALT levels of <2.5 _ ULN (P < .0001). In addition, HBeAg-positive Asian patients exhibited greater HBV DNA suppression with telbivudine than did patients from North America (P < .001) or other regions (P < .038).58
Valtorcitabine has also demonstrated potent suppression of serum HBV DNA in HBeAg-positive patients. In a phase I/II dose escalation study that evaluated valtorcitabine 300, 600, 900, and 1200 mg/day for 28 days, valtorcitabine 900 mg/day produced a mean reduction in serum HBV DNA level of 3.04 log10 copies/mL, representing a 99.9% reduction in viral load, after only 4 weeks of treatment.59 Valtorcitabine was well-tolerated in all patient cohorts, with a safety profile comparable to that of placebo.
The combination of telbivudine and valtorcitabine is also being investigated on the basis of the results of preclinical studies in a woodchuck model of HBV that indicated a synergistic effect between these agents in the inhibition of HBV replication. A phase II clinical study has been initiated to evaluate valtorcitabine as part of a fixed-dose combination therapy regimen with telbivudine. This will be studied in patients with CHB unable to achieve optimal therapeutic response to telbivudine in combination with another single agent.
Tenofovir
(large phase III studies of tenofovir in HBV are ongoing)
Tenofovir disoproxil fumarate, an acyclic nucleotide reverse transcriptase inhibitor structurally related to adefovir, which is used in the treatment of patients with HIV infection, has been shown to be effective in patients with HIV and HBV coinfection, particularly patients who are lamivudine resistant.60, 61, 62 The anti-HBV effects of tenofovir and adefovir were recently compared in a study of 53 lamivudine-resistant patients with CHB. Thirty-five patients received tenofovir for 72-130 weeks, and 18 received adefovir for 60-80 weeks.61 Changes in HBV DNA levels were followed for the complete period of 48 weeks. After 48 weeks of treatment, 100% of the tenofovir-treated patients had HBV DNA levels below 105 copies/mL compared with only 44% of adefovir-treated patients (P = .001). Side effects were similar among the treatment groups. No evidence of phenotypic viral resistance was observed up to 130 weeks in the tenofovir-treated patients.
These data suggest that tenofovir might become an effective alternative for the treatment of patients with lamivudine-resistant HBV infection. In the absence of long-term safety and efficacy data, tenofovir should be reserved for patients with HIV and HBV coinfection who exhibit lamivudine resistance.
Conclusions
Although conventional antiviral and immunomodulatory drugs used for managing chronic HBV infection have provided clinical benefit, these agents have limitations on the basis of unpredictable or suboptimal suppression of HBV viral replication, variable rates of resistance, and poor tolerability in the case of IFN. Newly approved antiviral therapies such as entecavir and peginterferon alfa-2a that have demonstrated increased potency and low to minimal drug resistance will enable the reduction of viral loads even further, an effect that is expected to induce higher rates of HBeAg and HBsAg seroconversion and to have a significant influence on the ultimate morbidity and mortality associated with CHB. Evidence from phase III clinical trials indicates that peginterferons are effective for the treatment of CHB, with no risk of viral resistance. Entecavir has so far demonstrated an excellent resistance profile, which needs to be confirmed by ongoing long-term follow-up studies. The nucleoside analogs emtricitabine, clevudine, telbivudine, and valtorcitabine and the nucleotide analog tenofovir have also demonstrated potent efficacy and good safety profiles in patients with CHB. Clinical studies of these agents alone and in combination with other anti-HBV therapies are ongoing. Combination therapy theoretically might improve efficacy and/or reduce or prevent the selection of antiviral resistant HBV mutations.
|
|
| |
| |
|
|
|