 |
|
|
| |
Resistance Mechanisms of the K70E HIV-1 Reverse Transcriptase Mutant
to GS-9148 and Other NRTIs
|
| |
| |
Reported by Jules Levin
ICAAC Sept 17-20, 2007, Chicago
K White, J Ly, A Eastoak-Siletz, G Lafl amme, N Kutty, A Ray, T Cihlar, and M Miller
Gilead Sciences, Inc., Foster City, CA, USA
INTRODUCTION
GS-9148 (Fig.1) is a novel structural analog of dAMP that acts as a nucleotide HIV reverse transcriptase (RT) inhibitor (NRTI)1
Both GS-9148 and its phosphonoamidate prodrug, GS-9131, exhibit potent activity in vitro against a broad range of NRTI-resistant HIV clinical isolates, including those with TAMs, M184V, or K65R2
In cell culture, GS-9148 selects for HIV-1 mutations in RT at codon 70, where K70N develops first and is then replaced by a triple mutant containing K70E+D123N+T165I3
K70E is present at a low frequency in NRTI-experienced patients (0.3%)3
K70R is a thymidine analog-associated mutation (TAM) involved in excision of NRTIs4
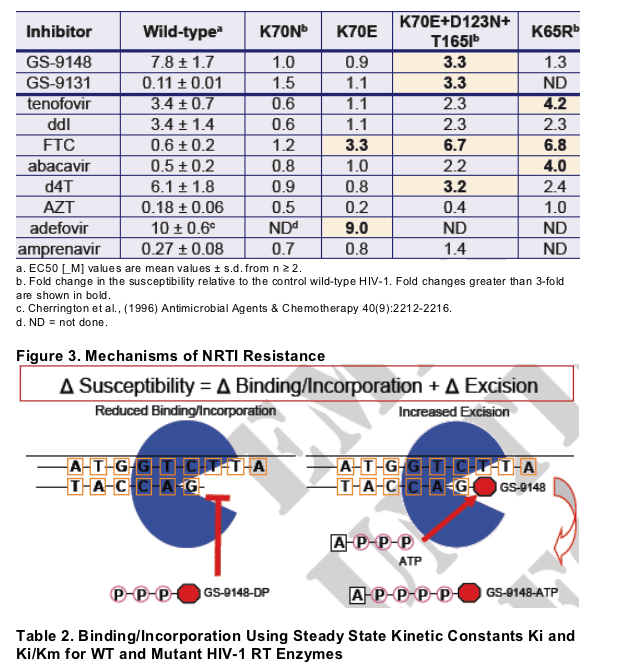
There are two major mechanisms of RT resistance to NRTIs: (1) decreased binding/incorporation, and (2) increased excision after incorporation
AUTHOR CONCLUSIONS
Viruses containing K70E+D123N+T165I, but not K70N or K70E alone, showed low-level decreases in susceptibility to GS-9148 (3.3-fold) and other NRTIs (2- to 3-fold), a 6.7-fold decrease to FTC, but were hypersensitive to AZT (0.4-fold) in cell culture
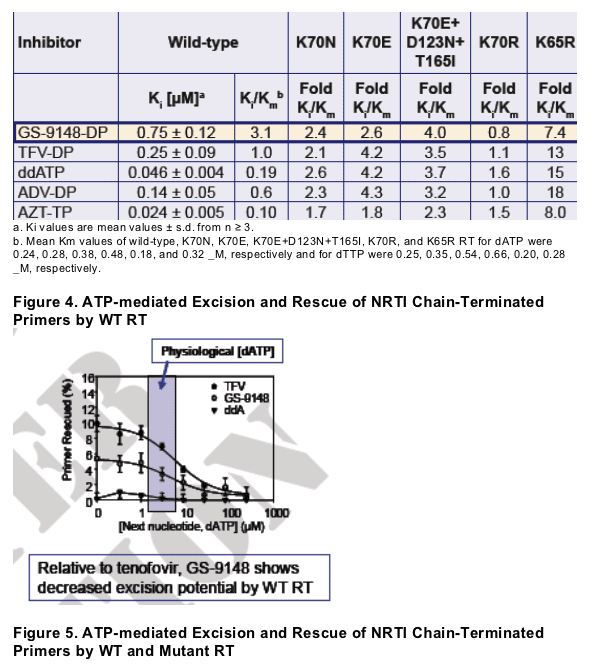
The progression of K70N to K70E to K70E+D123N+T165I mutant RTs corresponded to decreased binding/incorporation (increased Ki/Km) for GS-9148-diphosphate (2.4-, 2.6-, and 4.0-fold, respectively) relative to wild-type
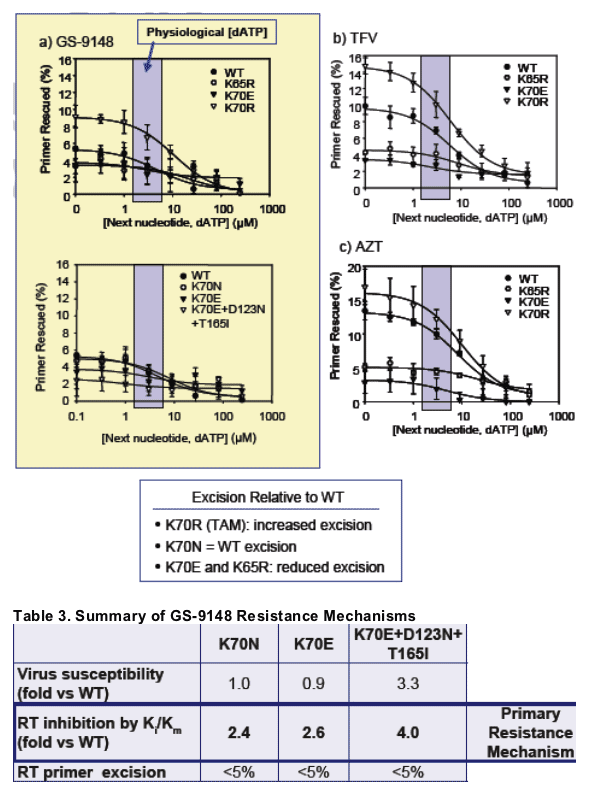
GS-9148 exhibited low levels of excision by both wild-type and K70E-containing RT
This mechanistic study confirmed the role of the K70E RT mutation in the resistance phenotype of GS-9148
For GS-9148 and other NRTIs, the resistance due to K70E, like K65R, was mediated primarily by decreased binding/incorporation of the inhibitors
The K70E RT mutant, like K65R and in contrast to the TAM K70R, showed reduced excision of NRTIs with measurable excision (tenofovir and AZT) relative to wild-type, that appears to counteract the incorporation defects for these NRTIs
These data suggest that the K70E mutant utilizes a resistance mechanism similar to that of K65R, but shows lower levels of resistance against NRTIs in cell culture and in biochemical assays
OBJECTIVE
To characterize the molecular mechanisms by which the K70N, K70E, and K70E+D123N+T165I mutations in HIV-1 reverse transcriptase cause reduced virus susceptibility to GS-9148 and other NRTIs
METHODS
Drug susceptibilities in cell culture were determined for wild-type (WT) and site-directed mutant HIV-1 strains in MT-2 cells using an XTT-based assay5
The steady state kinetic constants Ki and Km were determined for WT and mutant RT enzymes for the natural substrates dATP and dTTP and for the active metabolites of NRTI: GS-9148-DP, tenofovir-DP, ddATP (ddI), and AZTTP using a heteropolymeric DNA template5
The efficiency of excision of incorporated NRTI by the ATP-mediated excision
mechanism was determined using WT and mutant RT enzymes and increasing
concentrations of the next complementary nucleotide. Reactions were initiated
by the addition of 3.5 mM ATP and primers with NRTI excised were extended in
a rescue of polymerization assay with dNTPs and the Klenow polymerase6

References:
1. T Cihlar, A Ray, D Boojamra, L Zhang, H Hui, D Grant, K White, M Desai, N Parkin, and R Mackman. Abstr. 45, CROI 2006, Denver.
2. A Ray, J Vela, R Mackman, L Zhang, H Hui, R Pakdaman, A Carey, M Wright, G Rhodes, and T Cihlar. Abstr. 498, CROI 2006, Denver.
3. G. Lafl amme, D. Grant, K. White, K. Stray, C. Boojamra, L. Zhang, R. Mackman, A. Ray, M. Miller, and T. Cihlar , Abstr. H-1037 ICAAC 2007, Chicago.
4. V. Goldschmidt and R. Marquet. 2004. Int. J. Biochem Cell Biol. 36:1687-1705.
5. White, et al. 2002. Antimicrobial Agents & Chemotherapy. 46(11):3437-3446.
6. White, et al. 2005. AIDS. 19(16):1751-1760.
|
| |
|
|
|
|
|