 |
|
|
| |
In Vitro Antiviral Potency and Preclinical Pharmacokinetics of GSK364735
Predict Clinical Efficacy in a Phase 2a Study
|
| |
| |
Reported by Jules Levin
ICAAC Sept 17-20, 2007, Chicago
P.L. Golden1, S. Piscitelli1, S. Min1, S. Reddy1, B. Johns1, E. Garvey1, R. Ferris1, R. Hazen1, C. Edwards1, M. Woodward1, T. Yoshinaga2, A. Sato2, S. Iwashita2, K. Kadono2, T. Ohkawa2, K. Masuda2, K. Horie2, T. Fujiwara2, S. Palleja2
1GlaxoSmithKline, Research Triangle Park, NC; 2Shionogi and Company, Ltd., Osaka, Japan
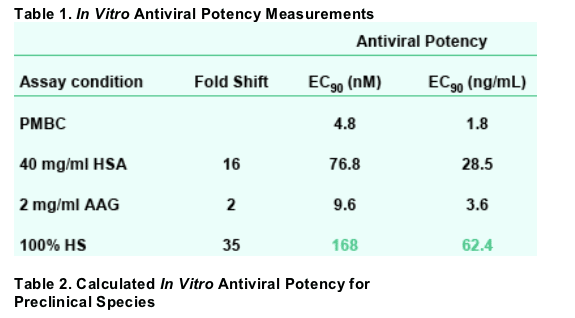
GSK364735 is a potent HIV integrase inhibitor, as demonstrated in a cellular assay. Its potency was attenuated in the presence of human serum albumin, AAG, and human serum; a potency shift of 35-fold is predicted in human serum.
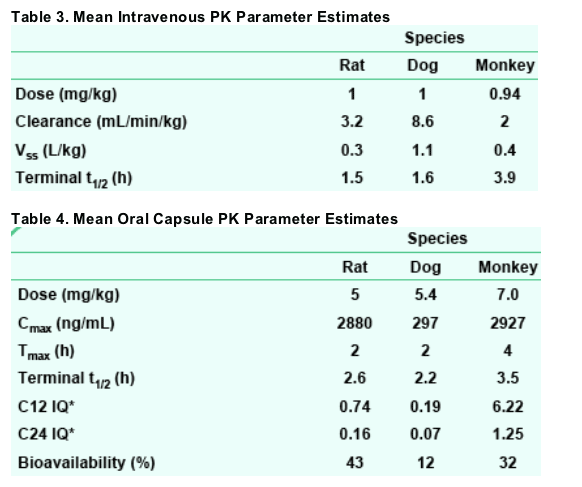
Following an IV bolus dose, the pharmacokinetic behavior in preclinical species is characterized by low clearance (ranging from 5 to 28% of hepatic blood flow; Davies and Morris, 1993) and moderate volume of distribution. Oral bioavailability of the potassium salt from a capsule ranged from 12% to 43% in preclinical species.
In addition to allometric scaling predictions (not shown) based on rat, dog, and cynomolgus monkey PK, the IQ ratios in cynomolgus monkey at 12 and 24 h after administration of a single capsule dose predict the potential for clinical efficacy of GSK364735 with once- or twice-daily dosing.
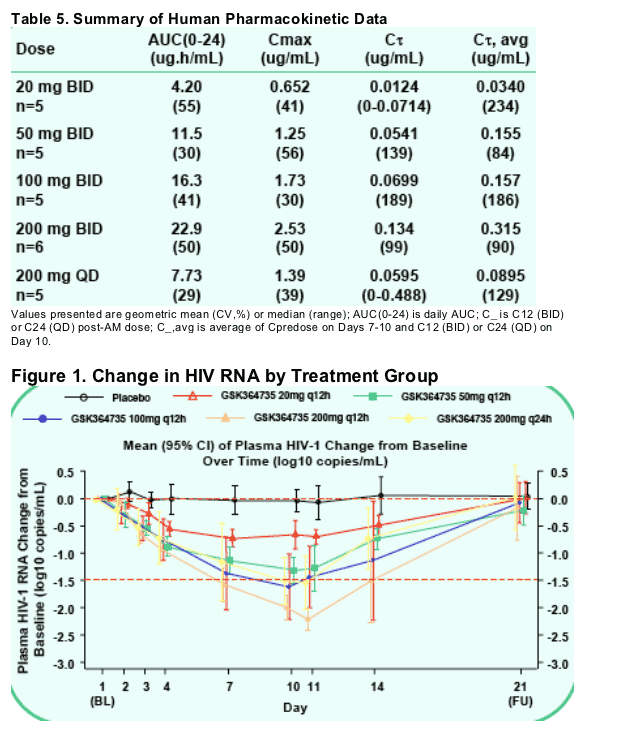
In HIV-infected individuals receiving GSK364735 for 10 days, total daily doses of 200 mg (either 100 mg q12h or 200 mg q24h) resulted in a greater than 1.5-log drop in HIV RNA. A 200 mg q12h dose resulted in a 2.2-log drop in mean RNA
after 10 days. While dosing with GSK364735C resulted in trough concentrations that exceeded the PA-EC90, there was wide intersubject variability and evidence of nonlinear absorption.
GSK364735 was successfully evaluated in two Phase I healthy subject studies and in a Phase II study in HIV-infected patients; however, adverse liver effects of
GSK364735 were recently observed in a long-term preclinical safety study in the monkey and preclude further development of the compound.
ABSTRACT
BACKGROUND: Among several factors, preclinical pharmacokinetics relative to serum-adjusted antiviral potency was used to select the lead naphthyridinone GSK364735, which was subsequently tested in HIV-infected patients in a Phase 2a study.
METHODS: Antiviral activity was measured in the presence and absence of human serum proteins. Intravenous and oral PK were studied in rat, dog, and monkey. Antiviral activity was evaluated in 32 HIV-infected patients who received GSK364735 as monotherapy for 10 days at doses of 20 mg, 50 mg, 100 mg, 200 mg each BID, 200mg QD, or placebo.
RESULTS: In PBMC cells, GSK364735's antiviral EC90 was 4.8 nM. When human serum was titrated and the effect extrapolated to 100% serum, potency decreased 35-fold. Thus, a therapeutic target of 168 nM was calculated. In PK studies, systemic clearance was low to moderate in all three species. At 12 h after an oral dose, plasma concentrations exceeded the protein-adjusted EC90 in rats and monkeys, while falling slightly below that target in dogs. Potassium salt oral bioavailability ranged from 9% (monkeys) to 45% (rats). In HIV-infected patients, GSK364735 demonstrated a dose-dependent decrease in HIV RNA
ranging from a mean -0.69 log decline at 20mg BID to -2.2 log decline at 200 mg BID. GSK364735 was well tolerated by HIV-infected patients.
CONCLUSIONS: Human dose projections based on preclinical potency and PK of GSK364735 predicted efficacy in HIV-infected patients with a low twice-daily dose. This was verified by a 2.2 log mean decrease in viral load observed in HIV-infected patients when administered GSK364735 at the highest dose tested in this trial for 10 days.
BACKGROUND
The use of combination antiviral therapy with HIV protease and reverse transcriptase inhibitors has resulted in significant improvement in AIDS-related morbidity and mortality. However, emerging multi-class drug resistant viral strains and long-term toxicities warrant development of new classes of antiretroviral
therapies.
GSK364735 (structure shown below) is a 2-metal-binding HIV integrase inhibitor; such integrase inhibitors preferentially block the strand transfer step in viral cDNA integration into the host chromosome.
GSK364735 was discovered and characterized by a triage lead optimization strategy that optimized structure-activity relationships for properties including antiviral potency, the effect of human serum and serum proteins on potency, preclinical pharmacokinetics, and preclinical oral bioavailability.
Following the selection of GSK364735 for development from a group of more than 200 compounds, its clinical PK and efficacy were characterized in studies in healthy subjects and HIV patients.
I. In Vitro Antiviral Potency
Methods
A five-day peripheral blood mononuclear cell (PBMC) assay used the HIV-1 strain Ba-L and PHA/IL-2 stimulated human PBMC cells, and quantitated virus in the cell supernatant via measurement of reverse transcriptase.
The effects of protein binding on inhibitor potencies were tested by adding 40 mg/mL human serum albumin (HSA), 2 mg/mL _1-acid glycoprotein (AAG), or human serum to the standard antiviral assay.
To approximate the effect of 100% human serum, serum was titrated from 0 to 20% and the effect was extrapolated to 100%, assuming a linear relationship between increasing serum concentration and decreasing inhibitor potency.
In vitro plasma protein binding was estimated by equilibrium dialysis against Sprague-Dawley rat, beagle dog, and cynomolgus monkey plasma.
RESULTS
II. Preclinical PK
Methods
Single-dose PK studies were conducted in male Sprague-Dawley rats, beagle dogs, and cynomolgus monkeys.
Intravenous bolus doses were administered in a DMSO:Solutol:0.05 M N-methylglutamine solution (1 mL/kg); oral doses were prepared with the GSK364735 potassium salt and an Avicel/sodium starch glycolate mixture filled into gelatin capsules.
Oral capsule doses were administered to animals that had been fasted overnight, with food provided at 4 h after dose administration. Quantitation of GSK364735 was performed by LC/MS/MS analysis of plasma samples.
Noncompartmental PK parameter estimates were obtained by nonlinear least squares regression analysis (WinNonlin Professional).
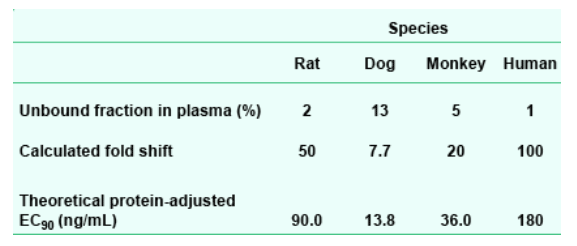
The inhibitory quotient (IQ; Hoefnagel et al., 2005) at 12 and 24 h was calculated for each species by dividing its plasma concentration (C12 or C24, respectively) after oral capsule dosing by its theoretical protein-adjusted IC90 (Table 2). While the original screening strategy used oral solution-dose exposures and human IC90 to calculate IQ (resulting in the data reported in the abstract), the current method accommodates each species' plasma protein binding-dependent pharmacokinetics.
All dose and exposure values are expressed as free acid equivalents.
RESULTS
III. Clinical PK and Efficacy
Methods
Phase 2a, multicenter, randomized, parallel, double-blind, dose ranging, placebo-controlled study.
Inclusion criteria included HIV-infected adults that were naïve or experienced to antiretrovirals, but naïve to integrase inhibitors and not currently receiving antiretroviral therapy.
Subjects had a CD4 count > 100 cells/mm3 and HIV RNA between 5000 and 300,000 copies/mL.
Subjects were randomized to receive one of the following treatments:
GSK364735 20mg bid, 50mg bid, 100mg bid, 200mg bid, 200mg qd, or placebo; all received the dose with food.
RESULTS
References
1. Hoefnagel JGM, Koopmans PP, Burger DM, Schuurman R and Galama JMD, Role of the Inhibitory Quotient in HIV Therapy. Antiviral Ther 10:879-892, 2005.
2. Davies B and Morris T. Physiological Parameters in Laboratory Animals and Humans. Pharm Res 10:1093-1097, 1993.
|
| |
|
|
|
|
|