 |
|
|
| |
An Open, Randomized, 2-Way Crossover Study to Investigate the Effect of Darunavir/Ritonavir on the Pharmacokinetics of Maraviroc in Healthy Subjects
|
| |
| |
Reported by Jules Levin
8th International Workshop on Clinical Pharmacology of HIV Therapy
Budapest, Hungary, 16-18 April 2007
S Abel1, C Ridgway1, J Hamlin1, J Davis1, R Mack2, V Sekar2
1Pfizer Global Research and Development, Sandwich Laboratories, UK
2Tibotec Inc, Yardley, PA, USA
Author's Summary of Findings
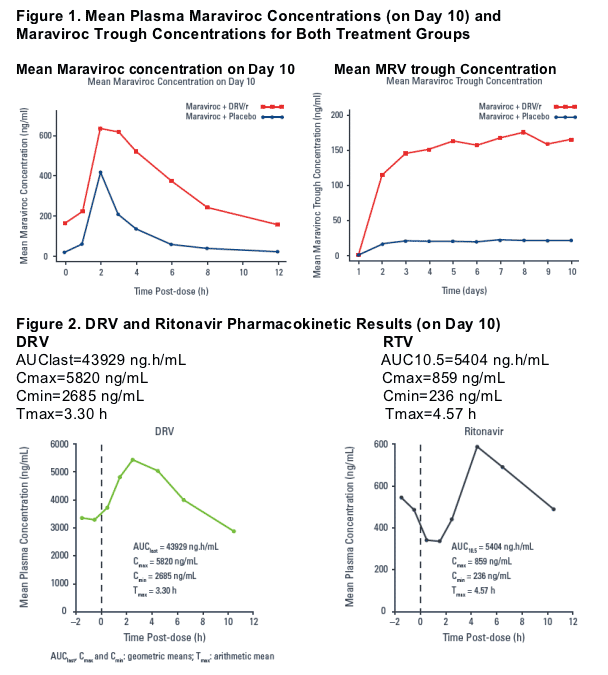
Analysis of maraviroc AUC12 and Cmax suggests that DRV/r increases exposure to maraviroc.
Visual inspection of individual trough concentrations suggests that steady state was achieved by Day 4.
There were few treatment related adverse events and all were of mild to moderate severity.
No clinically significant changes in laboratory safety tests, physical examinations, vital signs or ECGs.
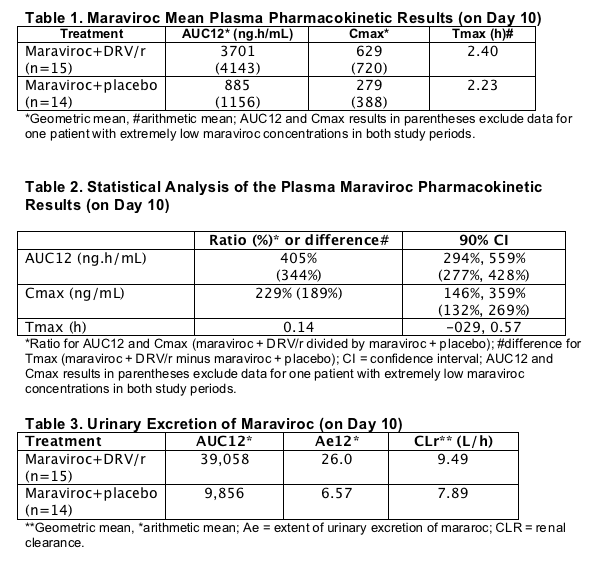
Pharmacokinetic parameters for DRV and ritonavir are similar to historical data for DRV/r (600 mg/100 mg BID) alone.6
Author Conclusions
There were no significant findings related to safety and toleration of the combination of maraviroc with DRV/r.
Co-administration of maraviroc with DRV/r leads to an increase in maraviroc exposure.
These data are consistent with previously reported effects for other PIs (except tipranavir/ritonavir) and other potent CYP3A4 inhibitors on maraviroc pharmacokinetics. Hence, it is recommended that a dose adjustment of 0.5-fold is made for maraviroc when co-administered with DRV/r.
Abstract
Background: The effect of darunavir/ritonavir (DRV/r; formerly TMC114/r) on the steady-state pharmacokinetics, safety and tolerability of maraviroc, a novel CCR5 antagonist for the treatment of HIV, was investigated in healthy subjects.
Methods: This was an open-label, randomized, placebo-controlled, 2-way crossover study in 12 healthy volunteers, aged 21-55 years. All subjects received 150 mg BID maraviroc on Days 1 to 9 and a single dose on the morning of Day 10. In addition subjects received DRV/r (600 mg/100 mg) or placebo BID on Days 1 to 9 and a single dose on the morning of Day 10. All subjects were fasted for at least 2 hours prior to, and for at least 1 hour post each maraviroc dosing. All DRV/r or placebo doses were administered 1.5 to 2 hours after maraviroc dosing and within 15 mins of completing a meal. Serial blood samples for determination of maraviroc plasma concentrations were collected on Day 10 from 0 to 12 hours post-dose. Each study period was separated by a minimum of 14 days. Maraviroc Cmax, Tmax and AUC12 were calculated on Day 10. Data were statistically analysed using an analysis of variance (ANOVA) appropriate for the study design.
Results: Geometric mean ratios (90% CI) for maraviroc AUC12 and Cmax (in the presence compared to the absence of DRV/r) were 405% (294%, 559%) and 229% (146%, 359%), respectively. There was no clinically significant difference in Tmax between the two treatment groups. Visual inspection of individual trough concentrations suggest that steady-state was achieved by Day 4. Co administration of maraviroc and DRV/r was well tolerated with no severe or serious adverse events.
Conclusion: Co administration of maraviroc with DRV/r leads to an increase in maraviroc exposure (AUC12 and Cmax). This data is consistent with previously reported effects for other protease inhibitors (except tipranavir/ritonavir) and other potent CYP3A4 inhibitors on maraviroc pharmacokinetics. Hence, it is recommended that a dose adjustment of 0.5-fold is made for maraviroc when co-administered with DRV/r.
INTRODUCTION
In the MOTIVATE 1 and 2 studies, maraviroc provided significantly greater virological suppression and CD4 cell count increases as well as fewer cases of treatment failure compared with placebo, when added to optimized background
therapy for antiretroviral therapy-experienced patients infected with CCR5-tropic HIV-1.1,2
An expanded access programme is ongoing to provide access to maraviroc for patients with limited or no other treatment options available to them due to resistance or intolerance.
Maraviroc is a CYP3A4 and P-gp substrate.3,4 Previous studies have demonstrated that its pharmacokinetics are affected by CYP3A4 and/or P-gp inducers and inhibitors. As such, a 50% dose reduction (i.e. 150 mg) is currently recommended in the presence of potent CYP3A4 inhibitors, including protease inhibitors (PIs; except for tipranavir/ritonavir).
Darunavir (DRV; developed by Tibotec) is a recently approved PI for use in treatment-experienced HIV patients.5 DRV is mainly metabolized by CYP3A4 and is given with ritonavir (a potent inhibitor of CYP3A4) to increase exposure at low doses.
When DRV and ritonavir (DRV/r) are co-administered in man, the net effect on metabolism appears to be inhibition of CYP3A4.
Given the potential for DRV/r to affect the pharmacokinetics of maraviroc, this study investigated the magnitude of any interaction to allow maraviroc dose-adjustment recommendations when these drugs are co-administered in clinical practice.
Objectives
To investigate the effects of DRV/r (600 mg/100 mg BID) on the steady-state pharmacokinetics of maraviroc (150 mg BID).
To investigate the safety and toleration of maraviroc when administered with DRV/r.
Methodology
The protocol was reviewed and approved by an independent ethics committee and all subjects gave written informed consent.
This study was an open, randomized, placebo-controlled, 2-way crossover study, in a group of 12 healthy subjects between the ages of 21 and 55 years.
For each subject, the study consisted of:
--screening visit up to 28 days before the start of dosing
--two treatment periods, each consisting of Day 0 until Day 10
--minimum wash-out period of 14 days between treatment periods
--follow-up visit 7-10 days after the last dose of study drug.
Subjects received maraviroc (150 mg) plus DRV/r (600 mg/100 mg) BID or placebo on Days 1-9 inclusive, with a single dose on the morning of Day 10.
All subjects were fasted for at least 2 hours prior to, and for at least 1 hours after, each maraviroc dosing. All DRV/r or placebo doses were administered 1.5 hours after maraviroc dosing and within 15 minutes of completing a meal.
Serial blood samples for determination of maraviroc, DRV and ritonavir plasma concentrations were collected on Day 10 from 0-12 hours post maraviroc dose.
Maraviroc Cmax, Tmax and AUC12 were calculated on Day 10. Data were statistically analysed using an analysis of variance (ANOVA) appropriate for the study design.
Cmax, Cmin, Tmax and AUClast for DRV and ritonavir were calculated on Day 10.
Safety and toleration data were collected throughout the study.
RESULTS
Analysis of maraviroc AUC12 and Cmax suggests that DRV/r increases the exposure of maraviroc.
There was no clinically significant difference in Tmax between the two treatment groups.
Visual inspection of individual trough concentrations suggests that maraviroc steady state was achieved by Day 4.
The amount of maraviroc in the urine was increased when co-administered with DRV/r; however, there was no clinically significant difference in CLR between the two treatment groups.
The range of DRV and ritonavir concentrations are consistent with those reported previously for subjects who received DRV/r 600 mg/100 mg BID (data not shown).66
There were no serious or severe adverse events. Treatment-related adverse events comprised of diarrhea, rash, dizziness, headache and nausea. All were of mild severity except for one rash reported as moderate severity.
There were no clinically significant changes in laboratory safety tests, physical examinations, vital signs or ECGs.
References
1. Nelson M et al. 14th Conference on Retroviruses and Opportunistic Infections, Los Angeles, USA, 25-28 February 2007. Presentation 104aLB.
2. Lalezari J et al. 14th Conference on Retroviruses and Opportunistic Infections, Los Angeles, USA, 25-28 February 2007. Presentation 104bLB.
3. Abel S et al. 6th International Workshop on Clinical Pharmacology of HIV Therapy, Quebec, Canada, 28-30 April 2005. Abstract 76.
4. Abel S et al. 7th International Workshop on Clinical Pharmacology of HIV Therapy, Lisbon, Portugal, 20-22 April 2006. Abstract 77.
5. Katlama C et al. AIDS 2007; 21:395-402.
6. Tibotec. Data on file, 2007.
|
| |
|
|
|
|
|