 |
|
|
| |
Inferring Virological Response from Genotype: With or without predicted phenotypes?
|
| |
| |
Reported by Jules Levin
16th Intl HIV Drug Resistance Workshop, Barbados, June 2007
André Altmann1, Tobias Sing1, Hans Vermeiren2, Bart Winters2, Elke Van Craenenbroeck2, Koen Van der Borght2, Soo-Yon Rhee3, Robert W Shafer3, Eugen Schülter4, Rolf Kaiser4,
Yardena Peres5, Anders Sonnerborg6, W Jeffrey Fessel7, Francesca Incardona8, Maurizio Zazzi9, Lee Bacheler10, Herman Van Vlijmen2, Thomas Lengauer1
1 Max Planck Institute for Informatics, Saarbrücken, Germany; 2 Virco BVBA, Mechelen; Belgium; 3 Stanford University, Stanford, CA, USA; 4 University of Cologne, Germany; 5 IBM Research, Haifa, Israel;
6 Karolinska Institute, Stockholm, Sweden; 7 Kaiser-Permanente Medical Care Program, San Francisco, CA, USA; 8 Informa, Rome, Italy; 9 University of Siena, Italy; 10 VircoLab, Inc., Durham, NC, USA
BACKGROUND, METHODS
Inferring response from genotype via the intermediate step of predicted phenotypes is often thought to have serious limitations. Here, we provide a retrospective comparison of approaches using either genotype or predicted phenotypes alone, or in combination.
Comparisons were based on two clinical datasets derived from 23,868 patients from Europe (EuResist) and California (Stanford-Kaiser). Response to antiviral therapy was dichotomized into success and failure according to the two EuResist standard datum definitions [1]:
In the classic definition a success is observed if the viral load (VL) is reduced below the limit of detection or more than two logs with respect to the baseline measurement after 8 weeks of treatment. Otherwise the therapy is considered a failure. In the alternative definition a sequencing event during an ongoing therapy marks a failure. A follow-up treatment that reduces the VL below the limit of detection is considered a success.
Computation of treatment scores
Four different types of inputs were used to compute treatment scores:
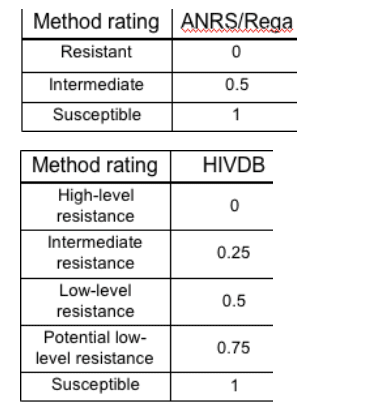
1. Expert algorithms: One number for each drug, representing the predicted activity of the drug for a given genotype. Activity was scaled between 0 and 1, with 0 indicating fully inactive drugs and 1 fully active drugs. Drugs not applied in a given treatment change episode were all assigned an activity level of 0. The mapping displayed by the tables on the right was used to translate the verbal rating given by the systems into a numeric value.
2. Predicted phenotypes: The VircoTYPE 4.0 [2] system is used to predict phenotypic resistance to the individual drug for a given genotype. Phenotypic resistance is given as the fold-change (FC) in log10(IC50). The FC in resistance was transformed into the interval [0,1], by using linear interpolation between the lower and upper cut-off (right figure). Results based on this input could be improved by using the relation between FC and %loss of wild-type response.
3. Raw genotype: A 0/1 representation of the genotype that indicates the presence (1) or absence (0) of a specific mutation. The list of mutations suggest by the International AIDS Society [3] was used to derive this representation. The given treatment was encoded in the same way. A 1 indicates the presence of a drug within the regimen, and 0 its absence
4. Hybrid: predicted phenotypes combined with either expert algorithm or raw genotype
Regimen scores for the "traditional" inputs (1 & 2) were derived from individual drug scores by summation. For comparison, specific weights were computed for each drug using support vector machines (SVMs). SVMs were also used to compute regimen scores for inputs 3 and 4. Model selection and evaluation was performed by nested cross-validation. Results are shown using receiver operating characteristic (ROC) curves.
CONCLUSIONS
Traditional summation scoring of treatment regimens can be improved by fitting weights for each drug
Predicted phenotypes as inputs were among the most sensitive non-hybrid approaches
Combining information on genotype and predicted phenotypes can improve predictive performance
RESULTS
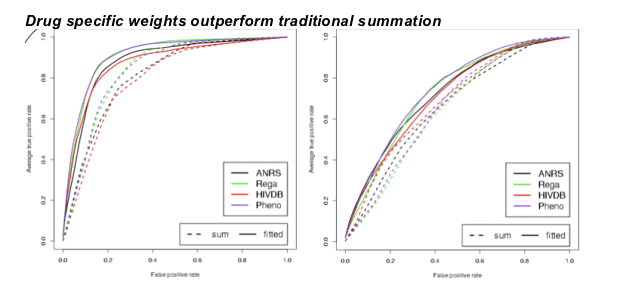
ROC curves obtained with 10-fold nested cross validation on the Stanford-Kaiser data using the alternative (left) and the classic definition (right). Dashed curves were computed using summation, and solid curves were computed using SVMs. Results obtained for both definitions show that SVM based combination (significantly) outperforms summation. Moreover, predicted phenotypes perform as well as expert-based approaches on both definitions.

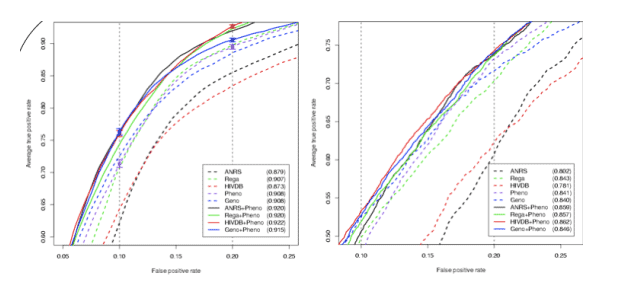
Partial ROC curves for single fitted approaches (dashed lines) and the hybrid inputs (solid lines) resulting from 10-fold nested cross-validation on the Stanford-Kaiser data (left plot) using the alternative definition. The AUC for the complete curve is given in the legend. Predicted phenotypes perform as well as the Rega tool and significantly outperform ANRS and HIVDB. Moreover, the hybrid inputs consistently outperformed the "traditional" (1 & 2) inputs. For confirmation, models from Stanford-Kaiser data were used to predict the completely separate EuResist data (right plot). The results are qualitatively identical, but the overall sensitivity is decreased. Using the classic definition the benefit of the hybrid encoding is less visible. However, predicted phenotypes are still among the most sensitive approaches.
|
| |
|
|
|
|
|