 |
|
|
| |
Safety and antiviral activity of BI 201335, a new HCV NS3 protease inhibitor, in treatment-naïve patients with chronic hepatitis C genotype 1 infection given as monotherapy and in combination with peginterferon alfa-2a (P) and ribavirin (R)
|
| |
| |
Reported by Jules Levin
AASLD Nov 4 2008 San Francisco, CA
Manns, Michael P.1; Bourliere, Marc2; Benhamou, Yves3; Pol, Stanislas4; Bonacini, Maurizio5; Berg, Thomas6; Trepo, Christian7; Wright, David8; Calleja, Jose L.9; Steinmann, Gerhard10; Huang, David B.11; Mikl, Jaromir11; Kukolj, George12; Stern, Jerry O.10, 11
1Medizinische Hochschule Hannover, Zentrum Innere Medizin, Hannover, Germany; 2Hopital Saint Joseph, Marseille, France; 3Hopital Pitie Salpetriere, Paris, France; 4Hopital Cochin, Paris, France; 5California Pacific Medical Center Research Institute, San Francisco, CA, USA; 6Charite Berlin Campus Virchow-Klinikum, Berlin, Germany; 7Hopital Hotel Dieu, Lyon, France; 8Central Texas Clinical Research, Austin, TX, USA; 9Hospital Universitario Puerta de Hierro, Madrid, Spain; 10Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany; 11Boehringer Ingelheim Pharmaceuticals Ridgefield, CT, USA; 12Boehringer Ingelheim (Canada) Ltd, Laval, Canada
AUTHOR DISCUSSION AND CONCLUSIONS
BI 201335 induced a pronounced virologic response typically within 2 to 4 days of initiation of monotherapy (from Jules: median maximal -4.2 logs in high dose group of 240 mg given for 14 days monotherapy)
-- 96.2% of patients achieved >2 log10 viral load decline during 14 days of BI 201335 monotherapy
-- 100% of patients in the 48 mg, 120 mg and 240 mg dose groups who received BI201335 achieved >2.8 log10 drop in VL during the first few days of monotherapy
-- The initial virologic response during monotherapy was not maintained; VL breakthrough occurred in the majority of patients by Day 14
BI 201335 given once daily as monotherapy for 14 days, followed by combination therapy with PegIFN/RBV for an additional 14 days, was well tolerated at all dosage levels among treatment-naïve patients
With the exception of an increased incidence of unconjugated hyperbilirubinemia at higher dosage levels, AEs were generally mild to moderate in severity and not considered to be related to study drug, and no patients discontinued monotherapy because of study drug_related AEs
-- AEs observed were commonly associated with PegIFN/RBV treatment
The results of this analysis support further study of BI 201335 once daily as combination therapy for treatment-naïve patients with chronic HCV infection
ABSTRACT
Background: BI201335 is a HCV NS3 protease inhibitor (EC50 of 3-6 nM). A multiple rising dose study evaluated safety and antiviral activity in treatment-naive patients (pts) with chronic HCV genotype-1 infection as monotherapy for 14 days followed by triple combination therapy with P+R for an additional 14 days.
Methods: 34 patients (France, Germany, Spain, USA) with a Metavir fibrosis score of 0-3 and no prior therapy with any IFN or R were randomized (2 placebo:6 active) to 4 dose groups of once-daily (qd) BI201335: 20 mg (n=8), 48 mg (n=9), 120 mg (n=9), or 240 mg (n=8). BI201335 was given as monotherapy for 14 days. Pts with <1 log10 decrease in Day 10 viral load (VL) had BI201335 discontinued after Day 14. Pts with a ≥1 log10 decrease in Day 10 VL continued BI201335 on Day 15 and added P(180ug/week) + R(weight based) for triple combination therapy through Day 28. The primary endpoint was ≥2 log10 VL reduction at any time to Day 14. Plasma HCV-RNA levels were measured with Roche COBAS TaqMan (LLOQ 25 IU/mL).
Results:
33 pts were white, 1 was Asian, 27 were male, mean age = 48.9±11.1 years, mean body weight 79.1±17.5 kg, and median (range) baseline VL was 6.8 (4.7-7.7) log10. There were no significant demographic differences between dose groups. BI201335 was well tolerated. No pts discontinued treatment during monotherapy due to adverse events (AEs). AEs observed were typical for P+R. One serious AE, asthenia, occurred in the 20 mg dose cohort 6 days after initiating P+R. Rapid decline of VL was observed in all pts with maximal decline 2-4 days after starting BI201335. With the exception of 1 pt in the 20 mg cohort, all pts on BI201335 achieved > 2 log10 VL decline during the monotherapy period. Median (range) maximal reductions in VL during 14 day monotherapy for the 20 mg, 48 mg, 120 mg, and 240 mg groups were 3.0 (1.5-3.9), 3.6 (3.1-3.8), 3.7 (3.3-4.1), and 4.2 (3.6-4.8) log10, respectively. No significant change in VL was observed with placebo. VL rebound during treatment was seen in the first 14 days of monotherapy in a majority of patients from all dose groups. Population sequencing of the NS3/4A protease at baseline and rebound during treatment revealed selection of variants that confer in vitro resistance to BI201335.
Conclusion: BI 201335 as monotherapy for 14 days followed by combination with P+R for additional 14 days was well tolerated, and induced a strong and rapid antiviral response. The results support further study of BI201335 as a once-daily potent antiviral for treatment-naïve HCV patients.
INTRODUCTION
BI 201335 is a low molecular weight, peptidomimetic serine protease inhibitor (EC50 of 3-6 nM) that exhibits potent and specific inhibitory activity against the HCV serine protease (NS3/NS4) required for the maturation of HCV viral polyprotein
-- This mechanism of action suggests that BI 201335 in combination with other HCV therapies, has the potential to be an effective drug for the treatment of chronic HCV infection
The safety and pharmacokinetics of BI 201335 have been characterized in an escalating single dose study (trial 1220.3) and in a multiple rising dose, 21- to 28-day trial (1220.6) in healthy volunteers
In Trial 1220.3, treatment with BI 201335 was safe and tolerable in all single dose levels (4 mg to 1200 mg) studied
In Trial 1220.6, 21 to 28 days of treatment with BI 201335 was safe and generally well tolerated across all dose levels studied (20, 48, 120, and 240 mg once daily). No relevant clinical AEs or laboratory abnormalities were reported for the 20 mg and 48 mg groups
-- At higher doses an increased incidence of headache, gastrointestinal symptoms and unconjugated hyperbilirubinemia was observed
-- The 120 mg dose was associated with slight increases of indirect bilirubin in 3/6 subjects
-- The 240 mg dose was also well tolerated, but all 6 subjects experienced reversible indirect hyperbilirubinemia up to 2.5 x ULN
Subjects with Gilbert's polymorphism (GP) (UGT1A1*28) were also studied in Trial 1220.6. Of the 9 subjects with GP, all 5 subjects who were homozygous for Gilbert's polymorphisms experienced reversible indirect bilirubin elevations in the serum up to 4.8 x ULN compared to subjects (n=4) with heterozygous polymorphisms who experienced smaller elevations of indirect bilirubin
These data suggest an association between the UGT1A1 polymorphisms and unconjugated hyperbilirubinemia with high doses of BI 201335
Steady state pharmacokinetic parameters of BI 201335 from trial 1220.6 indicate that BI 201335 NA has a long half life (t1/2, ss = 22.3h-30.9h) which supports once daily dosing
METHODS
Trial 1220.2 is a phase 1b, multinational study that is evaluating the safety, efficacy, and PK data of multiple rising doses of BI 201335 in HCV genotype 1_infected, treatment-naïve (double-blind, placebo-controlled within dose cohorts), and treatment-experienced (open-label, in combination with pegylated interferon alpha-2a and ribavirin [PegIFN/RBV]; Manns MP, et al, AASLD 2008, Abstract 1882) patients
The current interim analyses review the safety and virologic response during 14 days of BI201335 monotherapy followed by an additional 14 days of treatment with BI 201335 in combination with PegIFN/RBV in treatment-naïve patients
Main criteria for eligibility include: no prior therapy with IFN, PegIFN, or RBV for acute or chronic hepatitis C infection; aged ≥18 years; HCV genotype 1 (1a, 1b, or mixed 1a/1b) confirmed by genotype analysis at screening; HCV Ab positive confirmation or detectable VL at least 6 months prior to screening; VL ≥100,000 IU/mL at screening; histologic evidence within 24 months prior to study enrollment of any degree of chronic necroinflammatory activity, or the presence of fibrosis (Ishak grade 1_4 or METAVIR grade 1_3)
Four dose levels were evaluated: 20 mg, 48 mg, 120 mg and 240 mg. For each dosage group, patients received either BI 201335 (n=6 or 7) or placebo (n=2) (Figure 1)
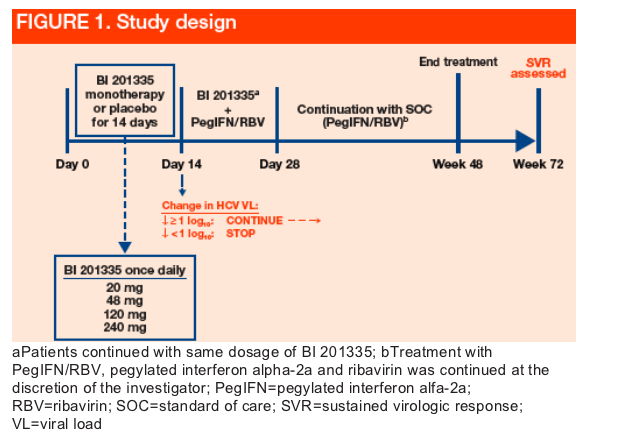
The effective target dose was selected based on PK models derived from preclinical data and from clinical PK data in trial 1220.6, a 4-week multiple rising dose study of BI 201335 in healthy volunteers, to meet a target median steady state trough plasma level (Cmin) of ≥17 ng/mL. This target exposure was reached with a single dose of 20 mg once daily, which was the first dose administered to treatment-naive patients
-- A data monitoring committee reviewed safety, efficacy, and PK data to ensure patients received a safe and virologically relevant dose
Plasma HCV RNA levels were measured using the Roche COBAS TaqMan (lower limit of quantification: 25 IU/mL; lower limit of detection: 10 IU/mL)
Patients with <1 log10 decrease in VL from baseline to Day 10 ended study at Day 14
Patients with ≥1 log10 decrease in VL from baseline to Day 10 received combination therapy of BI 201335 and standard doses of PegIFN alfa-2a/RBV from Days 15 to 28
-- After Day 28, at the discretion of the investigator, patients could continue to receive standard of care (SOC), PegIFN alfa-2a or 2b/RBV, up to Week 48
The primary efficacy endpoint was virologic response (VR), defined as a ≥ 2 log10 reduction in VL from baseline at any time point measured up to Day 14
Secondary efficacy endpoints included change from baseline in VL on Day 14; rapid virologic response (RVR), defined as undetectable VL (<10 IU/mL) on Day 28; end-of-treatment response (ETR), defined as undetectable (<10 IU/mL) VL at Week 48, and SVR
Primary safety endpoints include AEs, serious AEs (SAEs), and changes in laboratory abnormalities and test values over time
Secondary safety endpoints include the occurrence of AEs, by severity and by action taken with regard to test drug, and discontinuations due to AEs
Patients were monitored frequently for VL, treatment-emergent AEs, and laboratory abnormalities
--- VL was monitored on Days 1-4, 6, 10, 14 and 28
- VL was also measured on Day 21 in those patients with >1 log10 reduction in VL by Day 14
- For patients continuing with SOC, additional VL measurements were taken after Day 28 to monitor for early virologic response (EVR), end of PegIFN/RBV treatment response and sustained virologic response (SVR)
RESULTS
Baseline demographics
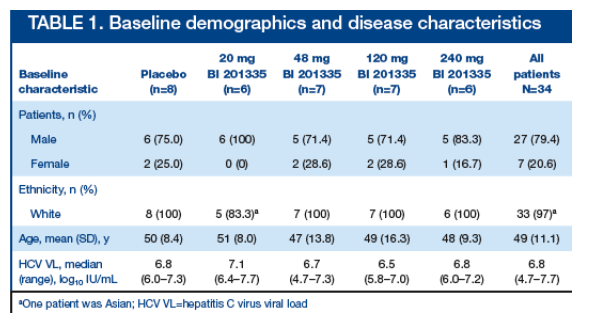
Thirty-four patients with no prior exposure to PegIFN or RBV were randomized to study drug or placebo within each of the 4 dose groups of once-daily BI 201335 or placebo. There were no significant differences in patient characteristics between study groups at baseline (Table 1)
Efficacy
With the exception of 1 patient in the 20 mg treatment group, all patients receiving BI 201335 (n=25, 96.2%) achieved the primary efficacy endpoint of >2 log10 VL decline during 14 days of monotherapy (Figure 2)
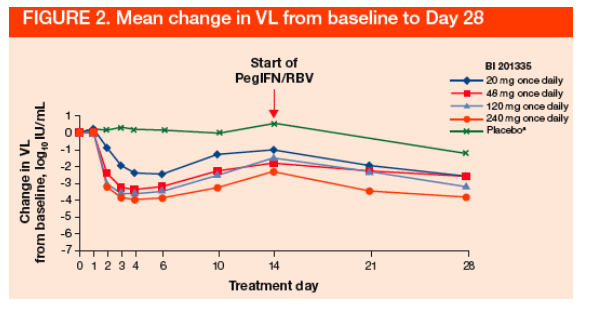
aVL not measured at Day 21 if <1 log10 VL reduction by Day 14 (8/8 patients in placebo group did not achieve <1 log10 VL reduction by Day 14); VL=viral load; PegIFN/RBV, pegylated interferon alpha-2a and ribavirin.
VL decline was rapid; maximal decline was typically reached 2 to 4 days after starting monotherapy (Table 2)

No significant change in VL was observed in patients in the placebo-control groups
Virologic breakthrough (defined as greater than 0.8 log10 increase in VL from baseline) during treatment was seen in the first 14 days of monotherapy in the majority of patients in all dose groups
Amplification of the NS3/NS4A protease segment followed by population-based sequencing of baseline and viral rebound samples demonstrated genotypic changes at specific residues within the NS3 protease domain consistent with resistant mutants that were previously characterized in in vitro resistance studies
Preliminary phenotypic characterization of the mutant NS3 protease domains in the context of the subgenomic replicon demonstrated shifts in the sensitivity to inhibition by BI 201335 consistent with previously characterized point mutants
Safety and tolerability
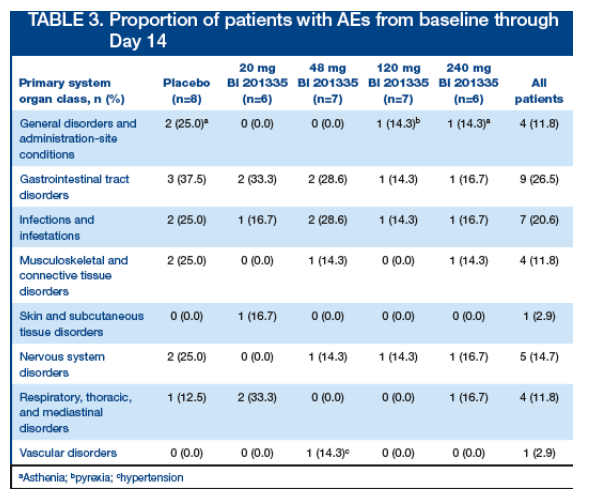
No dose-dependent increases in AEs were observed (Table 3)
Reported AEs were judged by the investigators to be unrelated to study drug and were typical of treatment with PegIFN/RBV, including fatigue, nausea, rash, headache, gastrointestinal tract disorders, and anemia
-- During the 14 days of BI 201335 monotherapy, diarrhea was reported in 1 placebo-treated patient and in 2 patients in the 48 mg BI 201335 group. There was no indication of any BI 201335 dose-related gastrointestinal effects
-- During the 14 days of BI 201335 monotherapy, no rash was reported in any study patient, however erythema was reported in 1 patient in the 20 mg BI 201335 group
-- Of note, during the triple combination treatment period (Days 15-28) with BI 201335 and PegIFN/RBV, a macular rash was reported in 2 patients: 1 in the 48 mg group and 1 in the 240 mg group. After 30 days following the completion of BI 201335 study treatment, new skin adverse events, including rash and erythema, continued to be reported on PegIFN/RBV follow-on treatment
One serious AE, asthenia, occurred in 1 patient in the BI 201335 20 mg dose cohort 6 days following initiation of therapy with PegIFN/RBV
No patients discontinued treatment during monotherapy because of AEs
No deaths were reported.
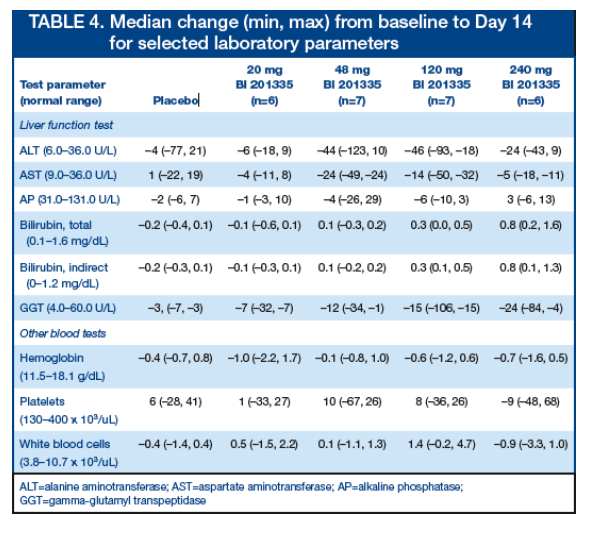
Changes in bilirubin were observed with increasing doses of BI 201335 (Table 4)
-- At higher doses, an increased incidence of unconjugated hyperbilirubinemia was observed
No other dose-dependent increase in clinical laboratory parameters was observed
REFERENCES
1. Viral hepatitis fact sheet, updated July 21, 2008. Centers for Disease Control and Prevention Web site.
http://www.cdc.gov/hepatitis/HCV/HCVfaq.htm#section1. Accessed September 15, 2008.
2. Dienstag JL, McHutchison JG. American Gastroenterological Association medical position statement on the management of hepatitis C. Gastroenterology. 2006;130:225-230.
|
| |
|
|
|
|
|