 |
|
|
| |
Phase 2 Study of Telaprevir Administered q8h or q12h with Peginterferon Alfa-2a or Alfa-2b and Ribavirin in Treatment-naïve Patients with Genotype 1 Hepatitis C: Week 12 Interim Results
|
| |
| |
Reported by Jules Levin
59th Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA, USA, October 31-November 4 2008
Xavier Forns,1 Patrick Marcellin,2 Tobias Goeser,3 Peter Ferenci,4 Frederik Nevens,5 Giampiero Carosi,6 Joost P Drenth,7 Koen De Backer,8 Rolf van Heeswijk,8 Tony Vangeneugden,8 Gaston Picchio,9 Maria Beumont-Mauviel8
1Liver Unit, University of Barcelona, Barcelona, Spain; 2Hopital Beaujon, Clichy, France; 3Klinikum der Universität zu Köln, Köln, Germany; 4Department of Internal Medicine III, Medical University of Vienna, Vienna, Austria; 5Department of Hepatology, University Hospital Gasthuisberg, Leuven, Belgium; 6Clinic of Infectious and Tropical Diseases, University of Brescia, Brescia, Italy; 7Radboud University Nijmegen Medical Center, Nijmegen, The Netherlands; 8Tibotec BVBA, Mechelen, Belgium; 9Tibotec Inc., Yardley, PA, USA
Premise
Telaprevir (TVR) is a potent and selective inhibitor of the hepatitis C virus (HCV) NS3⋅4A protease1 with demonstrated antiviral activity in both treatment-naïve patients and patients who failed prior therapy, including null responders to peginterferon (Peg-IFN) and ribavirin (RBV).2-7
In Phase 2 studies in treatment-naïve, genotype 1 HCV-infected patients (PROVE1 and PROVE2), a 24-week TVR-containing regimen (750 mg q8h) delivered a significant improvement in sustained virologic response rates over current 48-week therapy with Peg-IFN alfa-2a and RBV:
-- PROVE1: 61% vs 41% (P = 0.02)2
-- PROVE2: 69% vs 46% (P = 0.004).5
The objective of the C208 study (VX950-TiDP24-C208) is to explore the efficacy, safety, tolerability, and pharmacokinetics of TVR, administered as 750 mg q8h or 1125 mg q12h, in combination with Peg-IFN alfa-2a or Peg-IFN alfa-2b plus RBV (T/PR).
We report results from a planned interim analysis of C208 conducted at Week 12 of treatment.
METHODS
Overview
C208 is an ongoing, open-label, randomized, multicenter, Phase 2 exploratory study.
Adult, treatment-naïve patients with chronic genotype 1 HCV infection were enrolled. Patients with cirrhosis were excluded from the study.
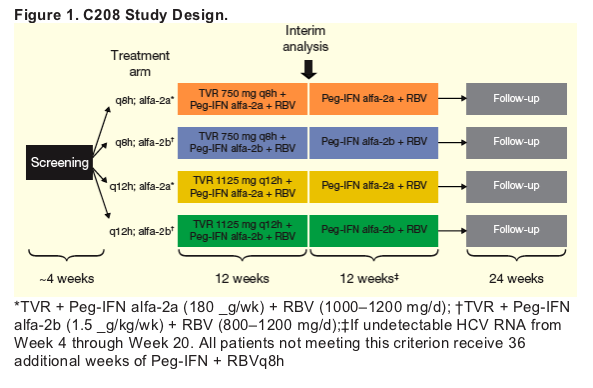
Patients are receiving 12 weeks of T/PR, followed by either 12 or 36 additional weeks of PR based on virologic response (Figure 1).
-- Patients with undetectable HCV RNA by Week 4 (rapid virologic response) through Week 20 stop treatment at Week 24.
Patients who did not meet this criterion are to receive 48 weeks of treatment.
-- Patients with HCV RNA >1000 IU/mL at Weeks 4, 6, or 8 are required to discontinue TVR dosing and complete 48 weeks of PR.
-- Patients with <2 log10 HCV RNA reduction at Week 12 are required to discontinue all treatment.
Efficacy Assessment
An interim intent-to-treat (ITT) analysis was performed and is reported here. Patients who discontinued the triple therapy were considered as non-responders in this analysis (NC=F).
-- This analysis includes all data up to the cut-off date, when all patients had reached Week 12 of treatment or had discontinued earlier.
The primary efficacy endpoints were the proportion of patients with undetectable HCV RNA at different timepoints throughout the first 12 weeks of dosing.
HCV RNA levels were measured using the COBAS TaqMan HCV RNA assay v2.0 (Roche Molecular Systems Inc., Branchburg, NJ, USA) (limit of detection [LOD]10 IU/mL; lower limit of quantification [LLOQ] 25 IU/mL).
Other Assessments
Virologic breakthrough (vBT; defined as a >1 log10 increase in HCV RNA from nadir, or HCV RNA >100 IU/mL in patients whose HCV RNA had previously become undetectable [<10 IU/mL]) was assessed.
Pharmacokinetic analysis of TVR was performed using noncompartmental methods for data from full pharmacokinetic profiles in a subset of patients.
Safety and tolerability were assessed by monitoring adverse events (AEs), vital signs, laboratory test abnormalities and electrocardiogram findings.
RESULTS
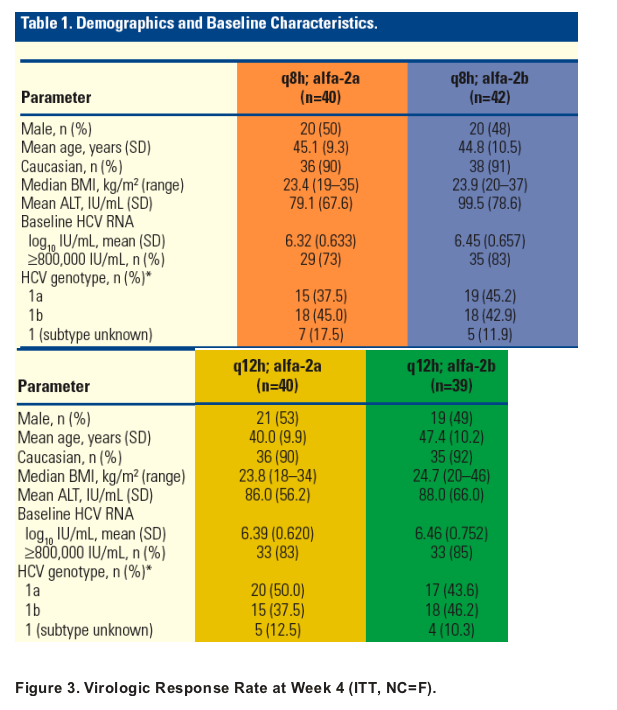
Demographics and Baseline Characteristics
Baseline characteristics were balanced across all groups (Table 1).
Efficacy Analysis
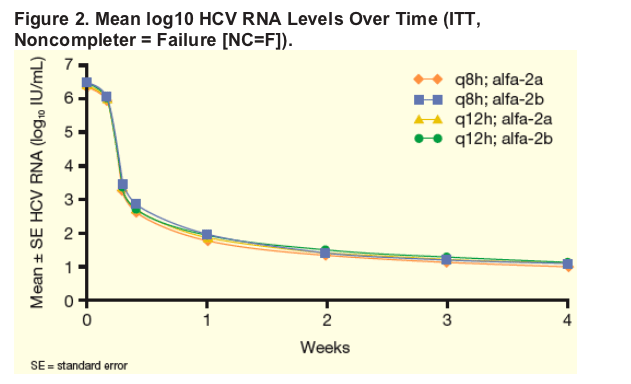
All treatment groups demonstrated similar rapid and potent suppression of HCV RNA from baseline to Week 4 (Figure 2).
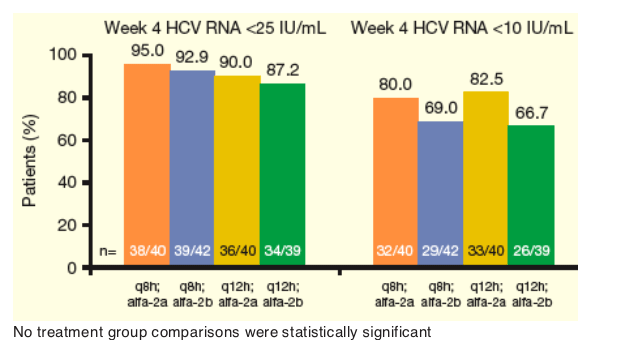
In all treatment groups, the majority of patients achieved undetectable and below LLOQ HCV RNA levels at Week 4: 67-83% achieved HCV RNA<10 IU/mL and 87-95% achieved HCV RNA <25 IU/mL (Figure 3).
-- Using a logistic regression model, no treatment group comparisons were statistically significant.
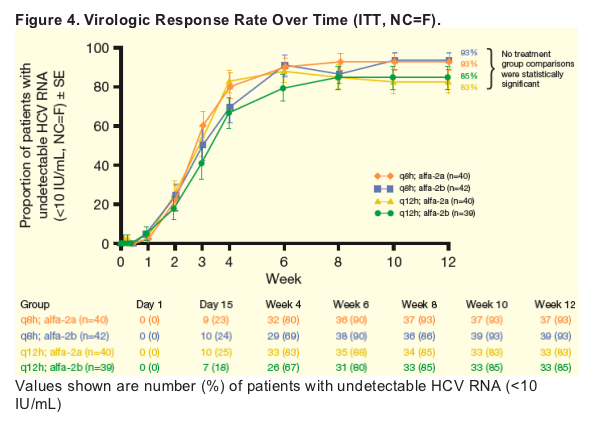
In all treatment groups, the majority of patients (83-93%) achieved undetectable HCV RNA at Week 12 (Figure 4).
-- No treatment group comparisons were statistically significant.
From Jules: So, in these two graphs there appears to be a difference, significant or not, between results at week 4 and week 12 in percent <10 Iu/ml. At week 4 the 3 times daily dose results are the same as the twice daily dose but at wek 12 there is a difference between 3 and 2 times daily dosing with 2 times per day having percent undetectable less than for 3 times per day. As to the reasons for this it is hard to sort out. It could be due to less potency with 2 times daily so resistance setting in, it could not be due to tis but to less adherence between weeks 4 and 12 to peg/RBV.
Virology
A total of 9 patients (5.6%) experienced vBT while on TVR across all arms of the study, of which:
-- 1 patient was in the q8h alfa-2a arm, 3 in the q8h alfa-2b arm.
-- 2 patients were in the q12h alfa-2a arm, 3 in the q12h alfa-2b arm .
-- vBT occurred within the first 4 weeks of treatment for 4 patients.
-- No patients achieved undetectable HCV RNA (<10 IU/mL) before meeting vBT criteria.
-- 8 patients were infected with HCV genotype 1a.
-- Using population sequencing, none of these 9 patients had variants with decreased susceptibility to TVR at baseline.
-- Variants with decreased susceptibility to TVR at the time of vBT were consistent with those previously described (V36M, R155K).
Two additional vBTs occurred after discontinuation of TVR, both in patients infected with genotype 1a.
One genotype 1a-infected patient, from the q8h alfa-2b group, had a R155K mutation at baseline by population sequencing. This patient achieved undetectable HCV RNA (<10 IU/mL) at Weeks 4 and 12.
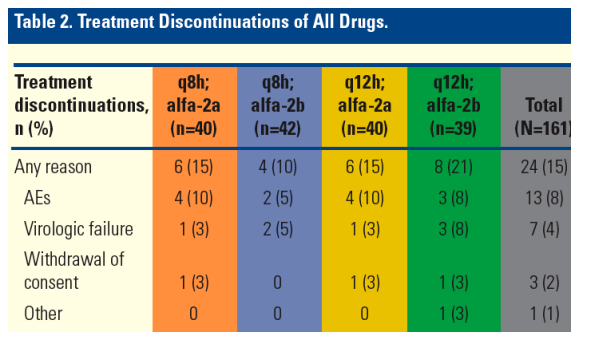
Treatment Discontinuations
The overall proportion of discontinuations of all drugs for any reason was 15% (Table 2).
Safety and Tolerability
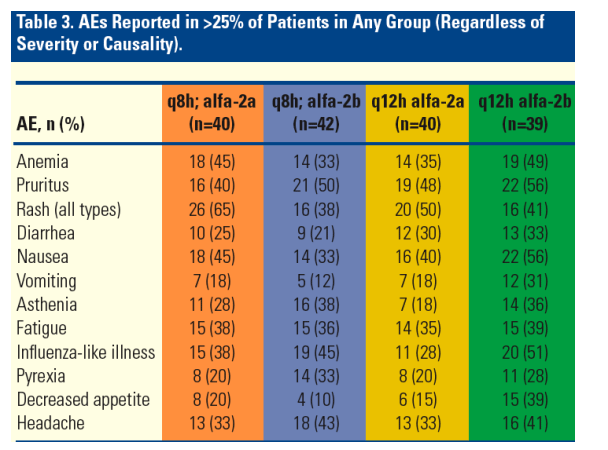
Adverse events (AEs) reported in this trial were consistent with those observed in earlier studies (Table 3).2,3,5
--- Overall, the incidence of any AE reported in >25% of patients in any group (regardless of severity and causality) was comparable between treatment arms.
Serious AEs leading to permanent treatment discontinuation were mainly due to rash- and anemia-related events.
Grade 3 rash occurred in 4 patients (2%). Grade 3 anemia occurred in12 patients (7.5%). The use of erythropoetin was restricted in the protocol.
-- Rash and anemia events improved upon cessation of treatment.
Laboratory Abnormalities
The majority of laboratory abnormalities were Grade 1 or 2 in severity.
Treatment-emergent Grade 3 laboratory abnormalities reported in >2 patients in the total study population were observed for low white blood cell count (n=32, 19.9%), uric acid (n=14, 8.7%), hemoglobin (n=15, 9.3%), lymphocytes (n=19, 11.8%) and neutrophils (n=33, 20.5%).
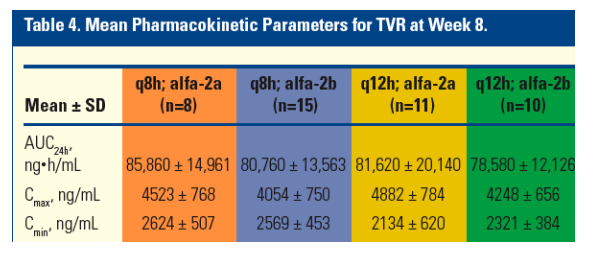
Pharmacokinetic Analysis (Substudy)
Total exposure to TVR (measured as AUC24h) was similar across all groups (Table 4).
On average, Cmax was higher and Cmin was lower for the q12h regimens compared with the q8h regimens.
Author Conclusions
The four telaprevir therapeutic regimens appear to be similar in efficacy and safety.
A high proportion of patients achieved undetectable HCV RNA(<10 IU/mL) with all four telaprevir-containing regimens.
-- These results are consistent with those in earlier studies.2,3,5
The rate of viral breakthrough was low (5.6%, n=9) and in line with previous studies.
AEs and treatment discontinuations were consistent with those reported in earlier telaprevir studies.
Total exposure (AUC24h) to telaprevir was similar across all four regimens.
Results of this interim analysis warrant further evaluation of a twice-daily telaprevir dosing regimen in treatment-naïve genotype 1 HCV-infected patients.
References
1. Lin K, et al. Antimicrob Agents Chemother 2004;40:4784-92.
2. McHutchison JG, et al. 43rd EASL 2008, Milan, Italy. 23-27 April 2008. Abstract 4.
3. Dusheiko GM, et al. 43rd EASL 2008, Milan, Italy. 23-27 April 2008. Abstract 58.
4. Poordad F, et al. 43rd EASL 2008, Milan, Italy. 23-27 April 2008. Abstract 1000.
5. Zeuzem S, et al. 59th AASLD 2008, San Francisco, USA. 31 Oct-4 Nov 2008. Abstract 243.
6. McHutchison JG, et al. 59th AASLD 2008, San Francisco, USA. 31 Oct-4 Nov 2008. Abstract 269.
7. Shiffman ML, et al. 59th AASLD 2008, San Francisco. USA, 31 Oct-4 Nov 2008. Abstract 1852.
|
| |
|
|
|
|
|