 |
|
|
| |
Cardiovascular Risk Markers Greatly Inflate Death Risk in People With HIV; interruptions increase risk
|
| |
| |
15th Conference on Retroviruses and Opportunistic Infections
February 3-6, 2008
Boston
Mark Mascolini
One coagulation marker and one inflammatory marker of cardiovascular disease inflated the risk of death from any cause in SMART trial participants interrupting antiretroviral therapy [1]. Principal investigator Lewis Kuller (University of Pittsburgh) told the 15th Conference on Retroviruses he'd never seen such heightened death risks with D-dimer in all his years of cardiovascular risk research.
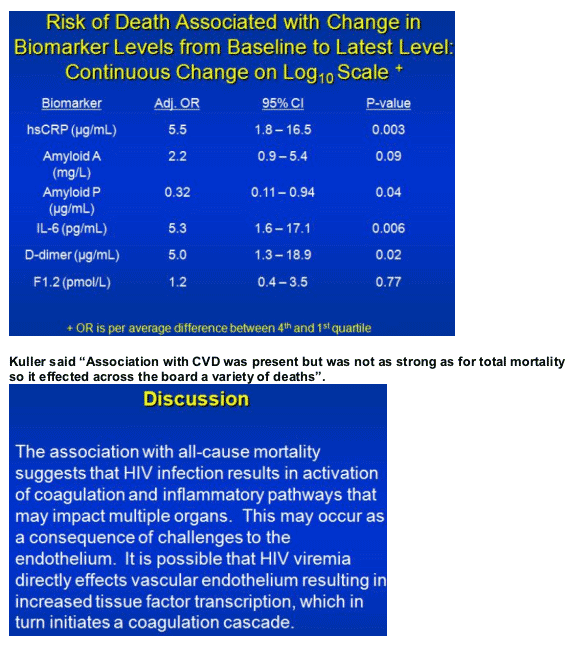
Kuller said "Association with CVD was present but was not as strong as for total mortality so it effected across the board a variety of deaths".
Analysis of a second treatment interruption trial, STACCATO, linked spiking viral replication after antiretroviral suspension to changes in key pro-inflammatory markers of endothelial dysfunction and found only partial reversal of these changes when people resumed treatment [2]. A third study correlated rebounding viremia during treatment breaks with T-cell activation and higher levels of two inflammatory markers of endothelial disease, including one of the markers singled out in STACCATO [3].
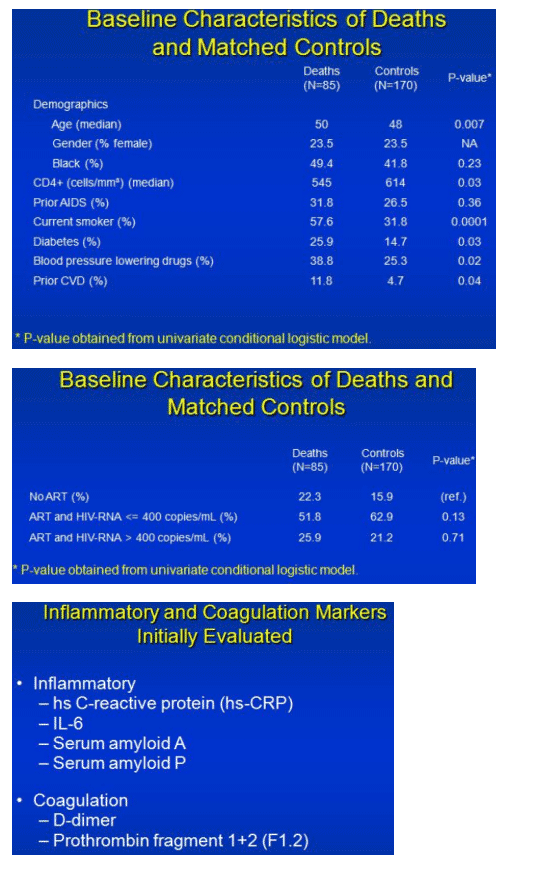
SMART randomized mostly treatment-experienced people to continuous antiretroviral therapy or to CD4-guided interruptions (stop if CD4s above 350, restart below 250). In the first 16 months 55 people in the treatment interruption group and 30 in the continuous therapy group died, only 7 of them from AIDS. For each of these 85 deaths, Kuller and colleagues selected two SMART study controls matched by country, age, gender, and approximate date of randomization. The people who died were significantly older (median 50 versus 48 years, P = 0.007) and included more current smokers (57.6% versus 31.8%, P = 0.0001), more people taking antihypertensives (38.8% versus 25.3%, P = 0.02), and more with a history of cardiovascular disease (11.8% versus 4.7%, P = 0.04).
Kuller evaluated them for four inflammatory markers (hs C-reactive protein (hsCRP), IL-6, serum amyloid A, and serum amyloid P) and two coagulation markers (D-dimer and prothrombin fragment 1 and 2). D-dimer is a fibrinolysis measure that signals thrombogenesis (clot formation). Overwhelming evidence implicates D-dimer as a powerful predictor of heart disease, according to Kuller.
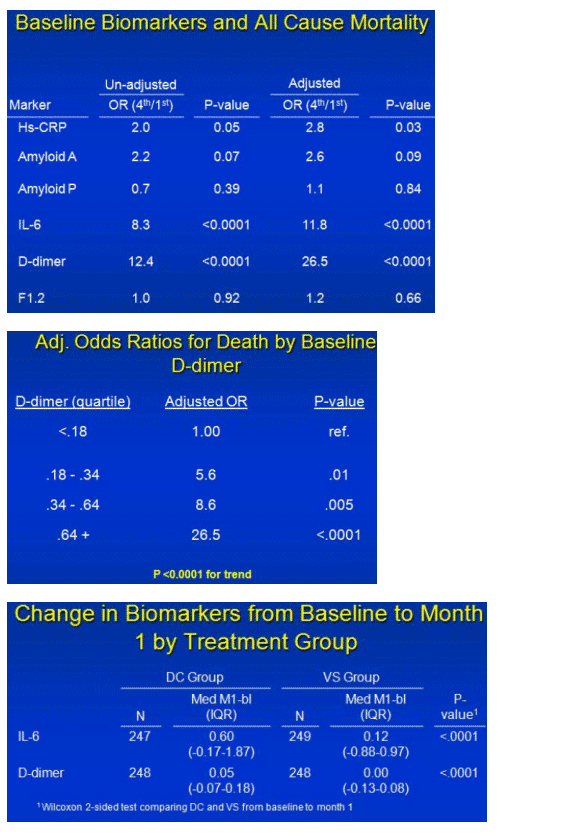
When people signed up for SMART, four markers already multiplied the risk of all-cause mortality from 2 to 26 times when Kuller compared patients with levels in the highest quartile to those with levels in the lowest quartile:
· hsCRP: adjusted odds ratio [OR] 2.8, P = 0.03
· Amyloid A: adjusted OR 2.6, P = 0.09
· IL-6: adjusted OR 11.8, P < 0.0001
· D-dimer: adjusted OR 26.5, P < 0.0001
The bloated risk with D-dimer, Kuller explained, is higher than reported in any other patient population. Compared with people in the lowest D-dimer quartile level (< 0.18 ug/mL), those in the second quartile (0.18 to 0.34 ug/mL) had a 5.6 times higher adjusted risk of death (P = 0.01), and those in the third quartile (0.34 to 0.64 ug/mL) had an 8.6 times higher risk (P = 0.005).
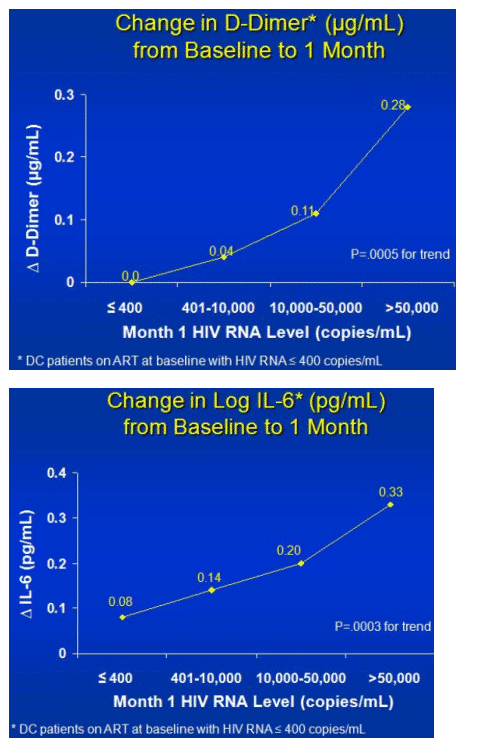
After 1 month in SMART, IL-6 and D-dimer levels rose significantly more in the interruption group than in the steady-therapy group. IL-6 concentrations soared 60% in break takers versus 12% in the control group (P < 0.0001), while D-dimer levels climbed 5% versus 0% (P < 0.0001). Changes in D-dimer levels correlated closely with viral load--the higher the load, the higher the D-dimer level (P = 0.0005 for trend). While Kuller saw no D-dimer change at 1 month in people whose viral load stayed below 400 copies, those with 401 to 10,000 copies had a 4% increase, those with 10,000 to 50,000 copies had an 11% increase, and those with more than 50,000 copies had a 28% increase.
Comparing fourth-quartile increases with first-quartile increases at the latest measurement, Kuller calculated significantly higher death risks with hsCRP (adjusted OR 5.5, P = 0.003), amyloid A (adjusted OR 2.2, P = 0.09), IL-6 (adjusted OR 5.3, P = 0.006), and D-dimer (adjusted OR 5.0, P = 0.02).
Kuller proposed that the link between all-cause mortality and higher marker levels means HIV infection activates coagulation and inflammatory pathways that affect multiple organs, not just the heart. Higher HIV load, he suggested, may directly affect vascular endothelium and increase tissue factor transcription, "which in turn initiates a coagulation cascade."
The investigators concluded that "elevated levels of D-dimer and IL-6 identify HIV-infected patients at very high risk of death. The magnitude of the association is clinically relevant and reasons for it require further study," as does the value of monitoring IL-6 and D-dimer levels in practice. Jumps in IL-6 and D-dimer during drug breaks "may explain, in part, the increased risk of all-cause mortality and cardiovascular disease" in the SMART interruption group.
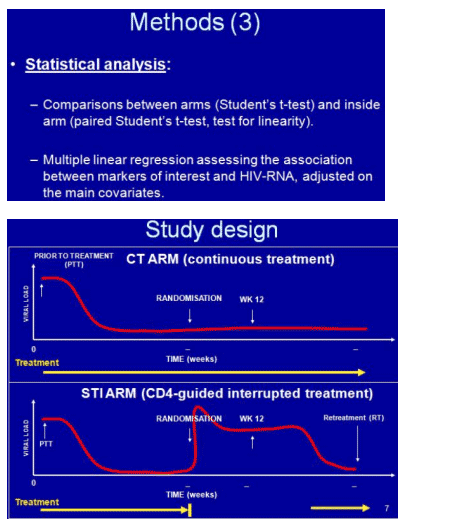
Hypothesizing that HIV replication results in endothelial activation and thus heightens the risk of heart disease, Alexandra Calmy (Geneva University Hospital) and STACCATO study colleagues analyzed activation markers in study participants who did or did not interrupt therapy [2]. The trial enrolled previously untreated people and after 6 months of therapy randomized those with an undetectable load and a CD4 count above 350 to continuous or off-and-on therapy (stop with more than 350 CD4s, restart with fewer than 350).
The analysis included 97 people from the treatment-interruption arm and 45 from the control group. Most people analyzed were women (62.1%) and young (mean 33.9 years + 8.1 standard deviation). Only 2% smoked. Baseline CD4 count and viral load stood at 286.7 cells and 40,000 copies.
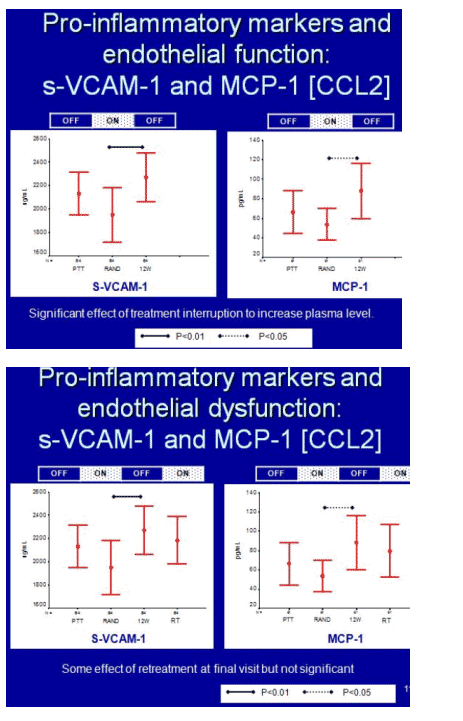
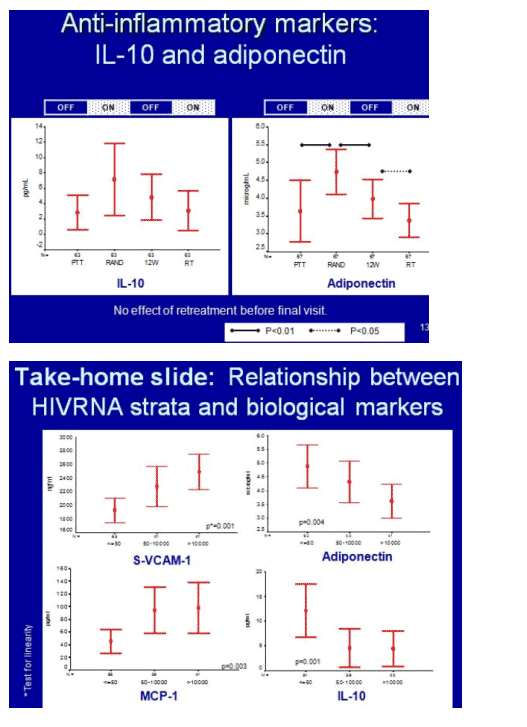
Levels of two pro-inflammatory markers of endothelial dysfunction, s-VCAM-1 and MCP-1, fell significantly when antiretrovirals began, then rebounded during the first drug break in interrupters (P < 0.01 for s-VCAM and P < 0.05 for MCP-1 for change during the first drug break). When interrupters resumed therapy, levels of both markers fell but did not return to the nadirs registered during the first treatment period. In contrast levels of two anti-inflammatory markers, IL-10 and adiponectin, rose during first-line therapy and fell during the first drug break. For adiponectin this rise (P < 0.01) and fall (P < 0.05) were statistically significant.
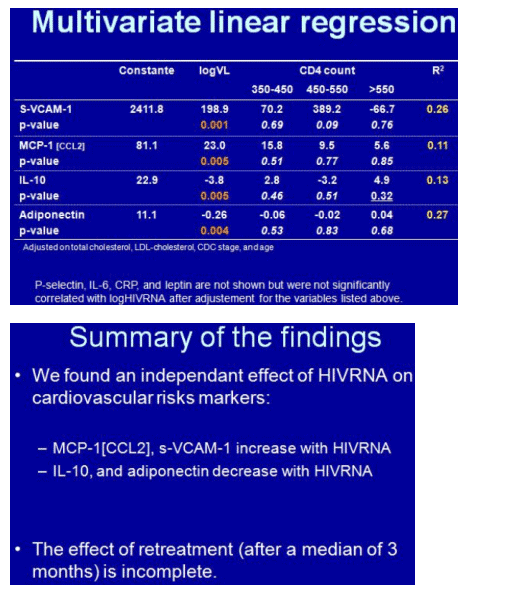
Levels of all four markers correlated significantly with viral load. For pro-inflammatory markers s-VCAM and MCP-1, higher viral loads meant high marker levels (P = 0.001 and 0.003). For anti-inflammatory markers adiponectin and IL-10, higher loads meant lower marker levels (P = 0.004 and 0.001).
A small retrospective comparison of treatment interrupters and continuously treated people by Wistar Institute and University of Pennsylvania researchers confirmed spiking pro-inflammatory markers during drug breaks [3]. These investigators also found jumps in T-cell activation during drug holidays.
This case-control study involved 52 people who kept their viral load below 500 copies for more than 6 months with antiretrovirals and had a load below 50 at the first follow-up point. Everyone had a CD4 count above 400 and a CD4 nadir no lower than 100. Group 1 consisted of 21 people analyzed at the start and end of a 6-week drug break; group 2 included 15 people analyzed at the beginning and end of 40 continuous weeks of treatment; and group 3 included 16 people analyzed when they started an open-ended drug break and when their rebounding viral load reached a set point.
In group 1 viral rebounds during the 6-week drug holiday resulted in significant jumps in CD3/CD95 ratio (P = 0.0022), CD8/CD38 ratio (P < 0.0001), and CD8/HLA-DR ratio (P = 0.001)--all results indicating stirred-up T-cell activation. In this group levels of the endothelial dysfunction markers s-VCAM-1 (P < 0.0001) and s-ICAM-1 (P = 0.003) climbed significantly during the drug break. Group 3 also had significant increases in both markers from the point when they stopped therapy to when their viral load hit a plateau.
Jumps in s-VCAM correlated positively with viral load after 6 weeks in group 1 (correlation = 0.55, P = 0.0098) and at the rebound set point in group 3 (correlation = 0.83, P = 0.0223). T-cell activation markers also correlated with s-VCAM and s-ICAM in group 1. In the steady-therapy group 2 the researchers saw no significant changes in either marker or correlations between activation markers and viral load.
The investigators argued that their findings demonstrate "the potential for a rapid and persistent endothelial stress response during short-term acute viremia."
References
1. Lewis Kuller and SMART Study Group. Elevated levels of interleukin-6 and D-dimer are associated with an increased risk of death in patients with HIV. 15th Conference on Retroviruses and Opportunistic Infections. February 3-6, 2008. Boston. Abstract 139.
2. Calmy A, Nguyen A, Montecucco F, et al. HIV activates markers of cardiovascular risk in a randomized treatment interruption trial: STACCATO. 15th Conference on Retroviruses and Opportunistic Infections. February 3-6, 2008. Boston. Abstract 140.
3. Papasavvas E, Azzoni L, Pistilli M, et al. Acute viral rebound as a result of structured treatment interruptions in chronically HIV-1-infected subjects: a pathway to cardiovascular risk? 15th Conference on Retroviruses and Opportunistic Infections. February 3-6, 2008. Boston. Abstract 964.
From Jules: the patient population in the first study had risk factors for CVD but in the second study they did not have these risk factors, it was a healthier group; there were no CVD endpoints in the second study.






|
| |
|
|
|
|
|