| |
Fluvastatin Inhibits Hepatitis C Replication in Humans- But Not Much
|
| |
| |
Am Jnl of Gastroenterology Apr 2008
Ted Bader, M.D.1,21Veteran's Administration Medical Center2University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma
".....Judged clinically, the effects of FLV on HCV RNA levels were modest. Fifty percent of patients experienced at least one lowering over 9-12 wk, but these reductions were short-lived for 2-5 wk. A curious phenomenon observed was that 4 patients had intermittent reduction in virus. Whether the rebound represents viral resistance or some adjustment by the host is unknown. In those with intermittent reduction, viral resistance would seem less likely as the reduction recurs at a later time...... FLV used as monotherapy in vivo showed suppressive effects on HCV clinically that are modest, variable, and often short-lived. These findings, along with other data suggesting synergism with α-interferon, support "proof-of-concept" for trials combining FLV with standard PI + R. Statins, and FLV in particular, appear to be safe for use in patients chronically infected with HCV. When needed, HCV patients should no longer be denied the hypocholesterolemic effects of statins."
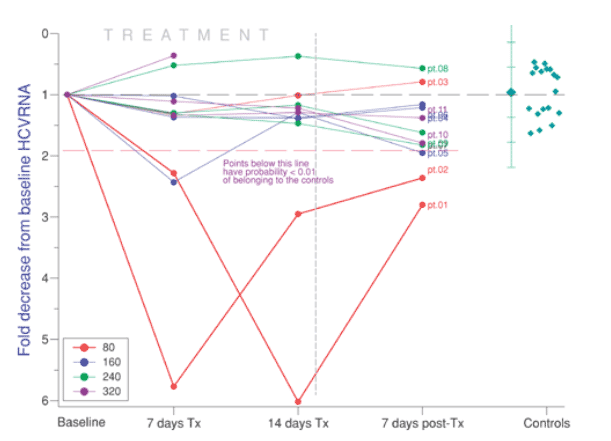
Figure 1. HCV RNA changes during 14 days of FLV at 80-320 mg/day. Two out of three patients taking 80 mg lowered HCV RNA significantly and then increased their viral load off drug. Patient 5 in the 160 mg dose group fell below the 0.01 probability line only at day 7 of treatment.
Abstract
BACKGROUND Hepatitis C viral (HCV) infection is the leading cause of death due to liver disease in the United States. Currently, pegylated interferon and ribavirin produce sustained viral remission in only 50% of patients. Additional agents are needed to increase the cure rate. In vitro experiments show strong antiviral effects of fluvastatin against HCV.
OBJECTIVES: To assess the safety and antiviral effects of fluvastatin in chronic HCV carriers.
METHODS: 31 veterans with chronic HCV were prospectively given oral doses of fluvastatin, 20 to 320 mg/day, for 2-12 weeks with weekly monitoring of HCV RNA and liver tests. Reductions of viral load (P < 0.01) versus a control group were considered suppressive.
RESULTS: With 80 mg a day or less, 11/22 (50%) patients responded by lowering HCV RNA. The first lowering occurred within 4 weeks (9/11, 82%). The greatest weekly change in HCV RNA level was a 1.75 log10 reduction. When lowered in responders, the viral load remained relatively constant for 2-5 weeks (7/9, 78%), or on the next test rebounded immediately to a non-significant change from, baseline (n = 2). Continued lowering of virus was seen in 2/19 (22 %) patients when the study ended. We found no evidence of liver tests worsening.
CONCLUSIONS: FLV used as monotherapy in vivo showed suppressive effects of HCV clinically that are modest, variable, and often short-lived. These findings support "proof-of-concept" for pilot trials combining fluvastatin with standard therapy. Statins and fluvastatin, in particular, appear to be safe for use in hepatitis C.
RESULTS
Study 1
Study 1 results are reported in Figures 1 and 2. All patients treated with the 80 mg ER dose responded: 2 of 3 patients taking 80 mg/day had a significant viral load reduction, while the remaining third patient in the 80 mg group improved his ALT. Regardless of dose and viral response, an improvement in ALT occurred in every patient (N = 3) who started the study with an abnormal ALT.
Study 2
Overall, in doses of 80 mg or less, 11 of 22 (50%) patients responded with at least one HCV RNA value lowered (P < 0.01) compared to untreated controls. The first lowering usually occurred within 4 wk (9/11, 82%), or came as late as 12 wk in 1 patient. The greatest weekly change in HCV RNA levels was a 1.75 log10 reduction (Fig. 3). When HCV RNA was lowered in responders, the viral load then remained relatively constant for 2-5 wk (7/9, 78%), or, in the next test, rebounded immediately to a nonsignificant change from the baseline (N = 2). Four of nine responders lessened their viral load intermittently. Continued significant lowering of virus was seen in 2 of 19 (22%) patients when FLV was stopped because of the duration of the study.
The only adverse reaction during either study occurred in a patient in study 1 who took the 320 mg dose for 5 days and developed nausea and diarrhea. This was judged as "possibly related to study drug" because symptoms of gastroenteritis have occurred at 640 mg/day FLV (13).
On day 41, patient 18 started an elevated ALT that rose to 851 IU/mL on day 68 (drug-held day 61ü\also stopped fish-oil capsules). During the rise in ALT, his viral load lowered from a range of 2-5 million IU/mL to 292,000 IU/mL. When his ALT was >3 X upper limit of normal (ULN), the total bilirubin remained ≦1.4 mg/dL; he reported increased energy and continued to work full-time. An IRB-approved drug rechallenge test with 20 mg FLV/day was negative. The rise in ALT was felt to be either because of his fish-oil capsules, or possibly was an attempt to clear HCV.
In patients without Gilbert's syndrome, the total bilirubin never exceeded 1.8 mg/dL. There was no unexplained muscle pain during the administration of FLV, and no creatine kinase value exceeded 10 X ULN.
DISCUSSION
The current methodology to evaluate new drugs for treatment of HCV is limited to documentation of dramatic log decreases in gross viral load. While this approach has strong specificity, it may lack sensitivity to detect other in vivo biological signals that suggest either host or viral therapeutic possibilities. Surely, the complex interaction of host-virus-drug must present other clues. For example, the acceptance of ribavirin as a valuable adjunctive drug was a historical quirk. It was developed in a time period before the availability of HCV RNA testing. If ribavirin were brought forth for evaluation of anti-HCV efficacy today, it would be quickly dismissed. Ribavirin as monotherapy has no discernable effect on viral load; this finding, combined with considerable universal toxicity, would eliminate it in phase I trials. However, a footnote to the long-standing consensus that ribavirin monotherapy does not affect viral load has recently been reported by Pawlotsky et al. (15) in that a significant antiviral effect occurs transiently on days 2 and 3 of the therapy, and not thereafter. Their viral load reduction for ribavirin qualifying for "significant effect" (-0.5-1.6 log10) is nearly identical to the current range reported here (-0.50-1.75 log10) for FLV.
We adopted the approach of fold change used by Nguyen et al. in the only reported study on the natural variation of HCV RNA in untreated HCV patients (11). We collected consecutive HCV RNA data for an untreated historical control group during the same year of the first study and observed viral load changes that matched Nguyen's data. Further, the changes seen in our control group have been further validated from an NIH trial with a 1-year placebo arm documenting HCV RNA variation (10) and Pawlotsky's control group (15). We wished to create a simple mathematical approach for visual evaluation. A statistical bar was set at the 99th percentile of the fold change in our untreated population; fold changes falling below this line (Figs. 1-3) have a 1% or less chance of belonging to a population of untreated controls. Displaying this 99% fold-reduction line on a scatter plot of all data points allows a discrimination of those points falling outside the expected range. Fold-reduction analysis appears to have an advantage over its exchangeable log reduction value when one visually examines more subtle viral load changes that include both extremely high and low viral loads. Moreover for practical use, it is far easier for a clinician to see that an HCV RNA value of 9,000,000 IU/mL falling to 3,000,000 IU/mL is a three-fold reduction and, thus, within natural variation (compared to the equivalent statement that a 0.5 log reduction occurred). The main disadvantage of fold variation, and why it should not replace log analysis, stems from the skewed results that occur with less than one-fold reduction. However, changes less than one fold represent background variation, and at that level, further investigation of an intervention would not be done.
Judged clinically, the effects of FLV on HCV RNA levels were modest. Fifty percent of patients experienced at least one lowering over 9-12 wk, but these reductions were short-lived for 2-5 wk. A curious phenomenon observed was that 4 patients had intermittent reduction in virus. Whether the rebound represents viral resistance or some adjustment by the host is unknown. In those with intermittent reduction, viral resistance would seem less likely as the reduction recurs at a later time.
The requirement that there should be a dose-response curve is not considered an absolute one in drug development (16). Increasing reductions in HCV RNA occur with mounting daily doses of α-interferon in the 1-10 million unit (MU) range; these effects flatten out at the 10-20 MU/day level; and above 30 MU/day, α-interferon becomes proviral and augments viral load.
How might FLV suppress HCV replication? Brown and Goldstein's proposed mechanism (see Introduction) is supported by in vitro observations. As such, one would expect to find a correlation between changes in serum LDL levels and viral load during FLV administration. We could not find any correlation between these variables (data not shown).
The disappearance of HCV during interferon treatment occurs in two kinetic phases. There is an initial rapid disappearance over 1-2 days and then a slower diminution that takes place over 2-8 wk. The initial phase is thought secondary to the clearance of virus within the plasma, while the second phase is because of the slower turnover of infected hepatocytes (17). If the model for interferon kinetics is applicable here, the inhibitory effects we report are consistent with suppression of virus within the plasma rather than intrahepatocyte reduction.
Two additional observations of HCV and LDL are germane. First, the structure and function of LDL produced by patients infected with HCV are significantly different from the LDL isolated from noninfected patients. These differences have been shown recently in vitro to alter immune system function (18, 19). Second, fluvastatin binds very strongly to LDL and alters the physicochemical characteristics of the lipoprotein to a greater degree than any other statin tested (20). Thus, we would hypothesize that FLV acts within the circulating plasma by binding to the LVP. The result of this FLV-LVP attachment may interfere directly with what we speculate may be the ability of the virus to modulate the immune system of the host. Also, the FLV-LVP complex may have more difficulty entering hepatocytes via the LDL receptor as the fluoride atom, after which the FLV is named, has a very large molecular size. As these targets may represent only minor hurdles to viral replication, this hypothesis might explain the modest viral load reduction seen. The dose-effect curve plateaus because there may be a limited number of binding sites for FLV on LVPs.
Inside the hepatocyte, FLV may bind to the lipid rafts that have been shown to be a possible location of HCV replication within the cell (21). The contribution of the depletion of prenylated proteins by reduction in mevulonate seen in vitro, the currently favored mechanism of action, remains to be determined as we could not find any correlation between LDL decline and viral load reduction.
Toxicity
In terms of ALT changes, two large retrospective studies reported a total of 340 HCV patients who were taking a statin. Compared to HCV non-statin users, neither study could find meaningful differences in ALT values (22, 23). Two prospective studies in HCV patients involving 20 mg/day atorvastatin and 20-40 mg/day rosuvastatin also reported no change in ALT (24, 25).
As ALT values unexpectedly improved for all patients in study 1 who started with an abnormal ALT (Fig. 2), we examined alternative ways to evaluate ALT changes within the context of statins and HCV. In our local retrospective HCV database, we had 13 patients with an abnormal ALT value when they initiated a statin; 12 improved their subsequent ALT value and 1 patient stayed unchanged. In the 12 patients that improved, using mean values, they decreased 60 IU/mL from a starting value of 140 IU/mL to 80 IU/mL. Forty-seven subjects had a normal ALT when the statin was started; 45 stayed within the normal range, while 2 had modest ALT elevations (<85 IU/mL); the latter 2 patients had evidence for concurrent heavy alcohol use, a known cause of aminotransferase elevation (26).
Depending how one evaluates ALT fluctuations, and whether one begins with an abnormal ALT, it can be said that FLV either improves ALT during treatment or makes no significant change. Larger prospective data sets will be needed to form the correct conclusion. We could not find any evidence that ALT worsens during FLV treatment. Drug toxicity is more accurately measured by bilirubin. No meaningful changes in total bilirubin were seen.
Looking ahead, there are three pieces of evidence that suggest fluvastatin might be additive or synergistic with α-interferon. Ikeda et al. have shown in vitro synergism against the replicon HCV virus when α-interferon and FLV are combined. They concluded, "We clearly demonstrated that combination treatment of α-interferon and FLV was an overwhelmingly more effective treatment, compared with the previous results for the combination treatment of α-interferon with ribavirin (1)." Second, a retrospective database examination of SVR with and without a statin by the Mayo Clinic showed that patients who unintentionally took a statin during PI + R therapy (N = 17) achieved an SVR of 82% versus 53% in controls (N = 34) (27). Third, after we were convinced that FLV was safe for use in patients with HCV, one of our genotype 1 patients on PI + R therapy, who had negative HCV RNA values at 12, 24, and 36 wk, broke through at week 46 of the therapy with three positive viral loads ranging up to 200,000 IU/mL. His compliance was certain as his platelet count had remained mildly lowered since the baseline. It is known that once a patient breakthroughs PI + R therapy, the likelihood of an eventual SVR is very small (28). We added FLV 80 mg/day and continued his PI + R regimen uninterrupted. HCV RNAs measured 12, 19, and 24 wk later have been negative. While continuing the same PI + R regimen, reversal of nonresponse after viral breakthrough has never been reported.
There were three limitations to our prospective studies. Only 1 of 31 patients was a female. Second, after completion of the second study, we felt the design of the dose titration arm was inadequate to evaluate the 40 mg and 60 mg doses as it did not allow any patient to start at those doses. Third, only the 80 mg dose is available as an ER; the prolonged serum half-life (3 h) of the ER formulation does not provide a straightforward comparison with the shorter-acting (1.2 h) 20-60 mg doses.
The failure of O'Leary et al. (24) or George et al. (25) to find any effect on HCV RNA with 20 mg of atorvastatin or 20-40 mg of rosuvastatin, respectively, in vivo might be considered to provide evidence against our report of FLV. However, our own testing of atorvastatin or rosuvastatin at those same doses also failed to show promise (data not shown).
CONCLUSIONS
FLV used as monotherapy in vivo showed suppressive effects on HCV clinically that are modest, variable, and often short-lived. These findings, along with other data suggesting synergism with α-interferon, support "proof-of-concept" for trials combining FLV with standard PI + R. Statins, and FLV in particular, appear to be safe for use in patients chronically infected with HCV. When needed, HCV patients should no longer be denied the hypocholesterolemic effects of statins.
BACKGROUND
Brown and Goldstein reported in 2003 that lovastatin (in large doses of 50 ā╩M/L) inhibited multiplication of the HCV replicon virus (3). They believe that the antiviral effect is another consequence of the principal effect statins possess by inhibiting HMG-CoA reductase, leading to lowered intracellular mevulonate. This inhibition, besides lowering LDL, also leads to reduction in geranylgeraniol, which ultimately leads to reduction in intracellular prenylated proteins. Prenylation is the process of attaching lipid moieties to protein, an operation essential for cell membrane formation, and a biochemical mechanism for which HCV apparently does not have encoded within its genome, and thus one which must be borrowed from the host cell (4).
Further evidence that low-density lipoprotein (LDL) might be critical for viral replication is suggested by numerous associations reported between LDLs and HCV. For example, the virus travels in the bloodstream as a passenger in lipo-viro-particles (LVPs) that consist of LDL, very low density lipoproteins, (VLDL) and apoB lipoproteins (5). The principle portal of entry for HCV into the hepatocyte is thought to be the LDL receptor (6). Significantly lower serum LDL levels are present in chronic carriers than in uninfected controls. Among the genotypes, genotype 3 patients have the lowest LDL, and more often have an associated hepatic steatosis; when genotype 3 is cured, LDL levels increase dramatically and the steatosis, if no other cause is present, disappears (7). Finally, higher pretreatment LDL values are a strong predictor of eventual sustained viral remission (SVR) (8).
The therapy currently approved by the U.S. Food and Drug Administration (FDA) is pegylated interferon and ribavirin (PI + R). In two trials, PI + R given for 48 wk resulted in SVR rates of 54-56% (8). Adverse effects of the therapy occur in nearly every patient and can be severe and life-threatening in 1-2% of patients (9). Additional agents are clearly needed.
Our study had two goals: evaluation of the safety of FLV in chronic carriers of HCV and observation for HCV viral load changes during monotherapy with FLV.
Study Design
All studies were approved by the University of Oklahoma Health Sciences Center Institutional Review Board. An investigational new drug (IND) license was required by federal law because of the fact that statins have a labeled contraindication for use in "active liver disease."
Two design principles were used as the basis for the trials. First, there is no placebo effect on plasma viral load or ALT during treatment of HCV (10). During licensure discussions with the FDA, placebo groups were not requested, required, or recommended. The scientific rationale for the basis of not using a placebo comes from Nguyen et al. (11) who demonstrated that HCV RNA levels measured periodically in untreated patients with chronic HCV usually stay within a two-fold change and never exceed a three-fold variation when measured over differing time intervals up to 3 months and diurnally. We confirmed the latter study findings by examining our own historical control group of untreated veteran HCV patients who were not alcoholic and were not on statins and who had more than one HCV RNA measurement during the year the first prospective study was done. Our control group had an absolute viral load of 2.76 ± 2.80 million IU/mL (mean ± standard deviation [SD]) and a 0.96 ± 0.41-fold variation between consecutive assays in 18 untreated HCV patients. These values are comparable to those of 29 patients in a placebo arm of a National Institutes of Health (NIH) ribavirin monotherapy trial that showed an absolute viral load of 3.4 ± 2.2 million units and a 1.1-fold variation in HCV RNA over 1 yr (10). This suggests that our study population is comparable to patient populations elsewhere.
A second design principle incorporated for study number 2 was the adoption of an "adaptive trial" protocol, a format that is particularly suited to pilot trials searching for dose efficacy. The goal of an adaptive trial is to evaluate interim findings and change doses during the study in order to gather the greatest amount of data while limiting drug exposure to the fewest patients. Decisions on dosage were made by the principal investigator in response to what effect different measurements were showing as the study evolved. Adaptive trials are increasingly thought to be superior to a rigid dosage protocol for phase I studies (12).
Prospective Study 1
The FDA approved dosage range for FLV is 20-80 mg/day. As normal volunteers using doses of 80-320 mg/day showed no significant adverse effects of FLV (13), the study was designed to test four oral doses of the extended release (ER) formulation of FLV (80, 160, 240, and 320 mg) given daily for 14 days with 3 patients at each dose level. Our rationale for such high doses was derived from an in vitro study. Ikeda et al. (1) used high ambient concentrations of FLV, 0.90 and 5.0 ā╩M/L, to show 50% and 99% suppression of HCV replicon virus, respectively, and these are equivalent to 389 ng/mL and 2,000 ng/mL. If these high levels were needed in the blood rather than the liver, we calculated that we could not achieve them. Maximum blood levels of FLV, after 13 days of therapy in normal individuals (daily oral dose, multiples of 80 mg ER tablets), have been reported as 61 ng/mL (80 mg), 162 ng/mL (160), 274 ng/mL (240), and 1,388 ng/mL (640) (13). However, most HCV replication occurs in the liver, and statins are highly concentrated in liver. FLV has a high first-pass absorption by the liver such that 50-80% of the drug is retained. Unfortunately, there are no data that relate oral doses or serum levels of any statin with the hepatocyte levels in humans. In rats, the comparison of a standard dose used in humans (1 mg/kg) versus an extremely high dose (19 mg/kg) produced the paradoxical result of a lower hepatocyte level of FLV, 9.0 ng/g liver versus 5.5 ng/g liver, respectively (14).
Our dosage plan was based on the in vitro data rather than on the meager serum-liver animal data; the result of which is that we postulated we needed to use higher oral doses of FLV than those approved for cholesterol. Even so, we anticipated the dosages might prove to be inadequate for the antiviral effect and thought we would at least develop more safety data for higher doses of FLV in hepatitis C patients.
Prospective Study 2
As study 1 appeared to show little or no benefit for doses of >80 mg, two arms were initiated to further study dosage levels around 80 mg. Arm 1 consisted of 11 patients who were started at 20 mg/day for 7 days and then titrated up each week to 40, 60, and 80 mg/day (only the 80 mg is available as an ER) for a total study period of 9 wk. Arm 2 began with 8 patients on 80 mg ER daily dose for total protocol duration of 12 wk. However, as data emerged suggesting 20 mg/day dose lowered viral load by a greater extent, some of the patients who were not responding to the 80 mg dose were lowered to 20-40 mg/day dose.
Study Participants
All 31 patients in the prospective studies were veterans, aged 44-61 yr, from the VA HCV clinic in Oklahoma City, Oklahoma. Twelve patients without cirrhosis were entered into study 1. Sixteen patients without cirrhosis and 3 patients with compensated cirrhosis were in study 2. The genotype distribution was 15 patients with genotype 1, 9 with genotype 2, and 7 with genotype 3. Nineteen participants were nonresponders to prior PI + R therapy, 3 were intolerant, and 9 were naive to PI + R.
Analytical Methods
HCV RNA levels were measured by the Roche COBAS TaqMan RNA real-time analyzer (Roche Diagnostics, Basal, Switzerland). ALT and total bilirubin tests were measured with the Beckman LXI Chemistry Analyzer (Beckman Coulter, Inc., Fullerton, California), with upper limits of 63 IU/mL and 1.2 mg/dL (20.5 ā╩M/L), respectively.
Statistical Methods
The a priori definition of a virological response to FLV was a five-fold reduction in the viral load compared to the pretreatment baseline value. In addition to this clinically defined response, a statistical bar was set at the 99th percentile of the fold change in an untreated HCV population based on the Gaussian distribution, with a mean and SD of the untreated group that were similar to those of McHutchison's paper. Thus, the values falling below this line (shown in Figures 1, 2, 3) have a 1% or less chance of belonging to a population of untreated controls, that is, they fall outside of the "noise" of variation seen in an untreated group.
Adjusting this line for multiple comparisons using the conservative Bonferroni correction (P = 0.0002) made no changes to the conclusions or to who was designated a responder. For simplicity, we show and discuss only the 99% threshold.
|
|
| |
| |
|
|
|