| |
Mechanisms of Neuronal Injury and Death in HIV-1 Associated Dementia
|
| |
| |
Current HIV Research, 2006, 4, 307-318
Marcus Kaul* and Stuart A. Lipton
Center for Neuroscience and Aging Research, Burnham Institute for Medical Research, La Jolla, CA 92037, USA
*Address correspondence to this author at the Center for Neuroscience and
Aging Research, Burnham Institute for Medical Research, 10901 North
Torrey Pines Road, La Jolla, CA 92037, USA; Tel: 001 858 646 3100 Ext.
3541; Fax: 001 858 713 6273; E-mail: mkaul@burnham.org
"Altogether, post mortem studies of human AIDS brains and experimental evidence regarding the pathologic mechanism of HAD indicate that synergy between inflammatory and excitatory pathways to neuronal injury and death may, at least in part, be shared with other CNS disorders including stroke, spinal cord injury and Alzheimer's disease. Newly developed therapeutic strategies for HAD will therefore likely benefit the treatment of several other neurodegenerative diseases and possibly vice-versa."
"Chemokine receptors allow HIV-1 to enter cells and as such are major potential therapeutic targets in the fight against HIV-1 infection and AIDS [94]. Inhibitors of CXCR4 and CCR5 prevent HIV-1 entry and are being assessed in clinical trials [94]. However, the benefit of chemokine receptor antagonists for HIV-associated neurological complications, although likely, remains to be shown.... All these findings support the hypothesis that selected beta-chemokines may represent a potential treatment modality for HAD."
"Given the substantial impact of inflammatory diseases on health in general, the pharmaceutical industry is currently developing p38 inhibitors for a variety of inflammatory- and stress-related conditions, such as arthritis, and this may expedite trials for CNS indications including HAD"
"Neuronal death by apoptosis appears to be one of the hallmarks of neurodegenerative diseases including HAD"
"Mood changes approaching the extent of disorders are one of many problems associated with HIV-1 disease.... Lithium has been suggested as a treatment for HAD...we have reported that the cytokine erythropoietin (EPO) may not only be effective in treating anemia but also for protecting neurons against inflammatory and excitotoxic injury, because it prevents NMDAR-mediated and HIV-1/gp120-induced neuronal death in mixed cerebrocortical cultures [23]. Since EPO is already clinically approved for the treatment of anemia, human trials of EPO as a neuroprotectant from HIV-associated dementia may be expedited [23]."
"While HAART has tremendously improved the treatment of HIV-1 infection and disease in the periphery, an effective pharmacotherapy for HAD (HIV-associated dementia) is still not available."
"HAART is unlikely to prevent the entry of HIV-1 into the CNS [66]. ......Consequently, as people live longer with HIV-1 and AIDS the prevalence of dementia might be rising and in recent years the incidence of HAD as an AIDS-defining illness has actually increased"..... The pathological features characterizing HIV-1 infection of the brain are......combined with selective neuronal loss"..... "Neuronal damage and loss has been observed in distinct brain regions, including frontal cortex"......"signs of neuronal death were not clearly associated with viral burden or a history of dementia"...."aging-associated amyloid accumulation with Alzheimer's-like neuropathology"..... "Altogether, it seems that a vicious cycle of immune dysregulation, inflammation and BBB dysfunction is required on the side of the host to allow sufficient entry of infected or activated immune cells into the brain and to permit neuronal injury"..... "Macrophages and microglia can be infected by HIV-1, but they can also be stimulated by factors released from infected cells."......"Certain HIV proteins, such as gp120, Tat and Vpr, may also exert some direct neurotoxicity"..... "toxic viral proteins among factors released from microglia and glutamate set free by astrocytes may all act in concert to provoke neuroinflammation and degeneration, even in the absence of extensive viral invasion of the brain"......"HAD might share the critical involvement of neuroinflammation and microglial activation with several other neurodegenerative diseases, such as Alzheimer's disease, Multiple Sclerosis, Parkinson's disease and Frontotemporal Lobe Dementia"......
"Neuropathological features that are similar to the findings in brains of AIDS patients.....all occur in the CNS of transgenic (tg) mice expressing HIV-1/gp120 [84, 122]. These gp120 tg mice develop significant behavioral deficits.....These findings suggest that the HIV-1 surface glycoprotein and engagement of the host's viral envelope receptors may well be sufficient to initiate neuronal injury and behavioral alterations.
Despite continuing progress in uncovering the pathologic processes, it remains a controversial topic how exactly HIV-1 infection provokes neuronal injury and death as well as neurocognitive and motor impairment
The hypothesis that HIV proteins can directly injure neurons without requiring the intermediary function of nonneuronal cells (microglia and/or astrocytes) is supported by experiments showing that viral envelope proteins are toxic in serum free primary neuronal cultures [91, 92] and in neuroblastoma cell lines
The HIV envelope protein gp120 interacts with several members of the chemokine receptor family (see above), and the direct form of HIV-induced neuronal injury may be mediated by chemokine receptor signaling in the absence of CD4
The hypotheses can be described as the "direct injury" hypothesis and the "indirect' or "bystander effect" hypothesis. These two hypotheses are not mutually exclusive
In addition to chemokines, MMPs and EAAs, HIV-infected or gp120-activated microglia also release inflammatory cytokines, including TNF-a and IL-1beta [107, 130]..... TNF-a can promote neurotoxicity
Experiments aimed at addressing the question of interactions between neurotoxins associated with HAD have revealed that TNF-a and HIV/Tat synergize to promote neuronal death, and this effect is prevented by antioxidants [104].....Indeed, we have found that antibody neutralization of TNF-a or inhibition of caspase-8 prevents the neurotoxicity
These findings suggest that inflammatory cytokines, including TNF-aand IL-1beta, may have important regulatory roles in HIV-associated neuropathology
Transgenic (tg) mice expressing HIV-1/gp120 in their CNS manifest neuropathological features that are similar to the findings in brains of AIDS patients, including reactive astrocytosis, increased number and activation of microglia, reduction of synapto-dendritic complexity, loss of large pyramidal neurons [122], and induction of MMP-2 [84]. In these tg mice, neuronal damage is ameliorated by the NMDAR antagonist memantine
Considering the evolving picture of HAD pathogenesis described above, several potential therapeutic strategies toattenuate neuronal damage are worth exploring. Among others, agents warranting consideration include NMDAR blockers, cytokines, chemokines, chemokine and cytokine receptor antagonists, p38 MAPK inhibitors, caspase inhibitors, and antioxidants (free radical scavengers or other inhibitors of excessive nitric oxide or reactive oxygen species)."
Abstract:
Infection with the human immunodeficiency virus-1 (HIV-1) and acquired immunodeficiency syndrome (AIDS) remain a persistent and even growing health problem worldwide. Besides its detrimental systemic effects on the immune system, HIV-1 seems to enter the brain very soon after peripheral infection and can induce severe and debilitating neurological problems that include behavioral abnormalities, motor dysfunction and frank dementia. Infected peripheral immune cells, in particular macrophages, appear to infiltrate the CNS and provoke a neuropathological response involving all cell types in the brain. Both viral and host factors, such as the viral strain and the response of the host's immune system, strongly influence the course of HIV-1 disease. Moreover, HIV 1-dependent disease processes in the periphery have a substantial effect on the pathology developing in the central nervous system (CNS), although the brain eventually harbors a distinctive viral population of its own. In the CNS, HIV-1 also initiates activation of chemokine receptors, inflammatory mediators, extracellular matrix-degrading enzymes and glutamate receptor-mediated excitotoxicity, all of which can activate numerous downstream signaling pathways and disturb neuronal and glial function. Although there have been
substantial improvements in the control of viral infection in the periphery, an effective therapy for HIV-1 associated dementia (HAD) is still not in sight. This article will review recently identified injurious mechanisms potentially contributing to neuronal death in association with HIV-1 disease and discuss recent and prospective approaches for therapy and prevention of HAD.
INTRODUCTION
The worldwide development of disease associated with infection by the human immunodeficiency virus-1 (HIV-1) and acquired immunodeficiency syndrome (AIDS) is alarming, with estimated numbers having grown from more than 35 million existing infections in 2001 to 38 millions in 2003, and more than 20 million deaths since 1981 [126]. Not only can HIV-1 destroy the immune system of its host and eventually lead to AIDS, the virus can also cause a variety of neurological problems that culminate in frank dementia.
AIDS-related opportunistic infections may affect the central nervous system (CNS) more often in the absence of treatment than in the presence of medication, but HIV infection itself can also induce a number of neurological syndromes [108]. Neuropathological conditions directly triggered by HIV-1 include peripheral neuropathies, vacuolar myelopathy, and a clinical syndrome of cognitive and motor dysfunction that has been designated HIV-associated dementia (HAD) [38, 42, 58, 112]. A mild form of HAD is termed minor cognitive motor disorder (MCMD) [27, 38, 58]. Interestingly, a high risk of neuropsychological impairment in HIV-1 infection seems to be indicated early on by anemia [88].
The mechanisms contributing to the development of MCMD and HAD remain incompletely understood, but the discovery in the brain of cellular binding sites for HIV-1, the chemokine receptors, and recent progress in understanding neuroinflammation and neural stem cell biology continue to provide new and surprising insights [43, 56, 58, 60, 66, 72, 95, 96]. The present article will review recent developments regarding the understanding of HIV-1's neurotoxic effect in the CNS and potential approaches for therapy and prevention of HAD.
HIV-1 INFECTION, AIDS AND NEUROLOGICAL DYSFUNCTION BEFORE AND SINCE THE INTRODUCTION OF HAART
HIV-1 productively infects macrophages and lymphocytes, first in the periphery and then in the brain, after binding of the viral envelope protein gp120 to one of several possible chemokine receptors in conjunction with CD4. Depending on the primary sequence of their gp120, different HIV-1 strains may use CCR5 (CD195) and CCR3, or CXCR4 (CD184), or a combination of these chemokine receptors to enter target cells [25, 48, 103].
Since most transmitted viruses use CCR5, deficiency in a functional receptor molecule (delta32-CCR5) can provide substantial protection against HIV-1 infection [79]. Some individuals who become infected though remain asymptomatic long-term and do not progress to AIDS have been found to express high levels of certain CCR5-binding beta-chemokines [106]. Again a few people never show seroconversion and seem to mount an unconventional, very effective humoral immune response that includes IgA antibodies against viral glycoprotein 41 (gp41) and IgG recognizing a CD4-gp120 complex [82].
Initially, the majority of severe neurological symptoms occurred in advanced stages of systemic HIV-1 disease and the prevalence of HAD was estimated to be 20-30% in individuals with low CD4 T cell counts [88]. In addition, anemia associated with HIV-1 infection presented itself as an early predictor for a high risk of neuropsychological impairment [88]. The introduction of highly active antiretroviral therapy (HAART) has increased the life expectancy of people infected with HIV-1 and resulted in an at least temporary decrease in the incidence of HAD to as low as 10.5% [87]. This transient effect attests to the point that the effects of HIV-1 infection in the brain should always be considered in conjunction with the systemic conditions and it is now widely understood that a peripheral infection and an associated immune response and inflammatory processes can influence all cell types in the CNS [16, 125]. Indeed, improved control of peripheral viral replication and the treatment of opportunistic infections continue to extend survival times, but HAART fails to provide protection from MCMD or HAD, or to reverse
the disease in most cases [21]. Although MCMD may be more prevalent than frank dementia in the HAART era, HAD constitutes a significant independent risk factor for death due to AIDS and it is assumed to be the most common cause of dementia worldwide among people of age 40 or less, [27]. Moreover, the proportion of new cases of HAD displaying a CD4 cell count greater then 200 μl-1 is growing [87], and another recent study found that in a group of 669 HIV patients who died between 1996 and 2001 more than 90% had been diagnosed with HAD as an AIDS-defining condition within the last 12 month of life [129]. This situation might at least in part be due to poor penetration into the CNS of HIV protease inhibitors and several of the nucleoside analogues, and distinct patterns of viral drug resistance in plasma and cerebrospinal fluid (CSF) compartments have also been observed [21, 60, 66]. While HIV seems to penetrate into the CNS soon after infection in the periphery, and then resides primarily in perivascular macrophages and microglia [36, 43, 52], current therapeutic guidelines for AIDS suggest to start HAART only once the numbers of CD4+ Tcells begin to decline. Since this might occur up to some years after peripheral infection, HAART is unlikely to prevent the entry of HIV-1 into the CNS [66]. Consequently, as people live longer with HIV-1 and AIDS the prevalence of dementia might be rising and in recent years the incidence of HAD as an AIDS-defining illness has actually increased [21, 56, 58, 60, 66, 76, 87]. Therefore, a better understanding of the pathogenesis of HAD, including viral and host factors, is urgently required in order to identify additional therapeutic targets for the prevention and treatment of this neurodegenerative disease.
NEUROPATHOLOGY OF HIV INFECTION AND DEVELOPMENT OF HAD
The pathological features characterizing HIV-1 infection of the brain are commonly referred to as HIV encephalitis (HIVE) and include widespread reactive astrocytosis, myelin pallor, microglial nodules, activated resident microglia, multinucleated giant cells, infiltration predominantly by monocytoid cells, including blood-derived macrophages, and decreased synaptic and dendritic density, combined with selective neuronal loss [85, 108]. Surprisingly, measures of cognitive function do not correlate well with numbers of HIV-infected cells, multinucleated giant cells or viral antigens in CNS tissue [1, 41, 85, 132]. In contrast, increased numbers of microglia [41], elevated tumor necrosis factor
(TNF)-a mRNA in microglia and astrocytes [130], evidence of excitotoxins [50], decreased synaptic and dendritic density, and selective neuronal loss [1, 85, 132] constitute the pathologic features most closely associated with the clinical signs of HAD. Neuronal damage and loss has been observed in distinct brain regions, including frontal cortex [31, 61], substantia nigra [114], cerebellum [46], and putamen [28]. The appearance of focal neuronal necrosis was reported for HAD brains earlier on [51], though more recently signs of neuronal apoptosis have been linked to HAD [3, 109]. In one study a good correlation was observed of HAD with cellular DNA fragmentation in basal ganglia detected by terminaldeoxynucleotidyl-transferase-mediated dUTP nick-end labeling (TUNEL) [115]. However, signs of neuronal death were not clearly associated with viral burden or a history of dementia [3]. The localization of apoptotic neurons is correlated with evidence of structural atrophy and closely associated with signs of microglial activation, especially within subcortical deep gray structures [3].
Since the introduction of HAART, however, HIV neuropathology is shifting. While the number of opportunistic infections appears to be reduced, the prevalence of HIV-associated encephalitis seems to be higher at autopsy [70].
Post mortem specimens from HIV patients who failed HAART showed even more encephalitis and severe leukoencephalopathy. Another post mortem study found increased macrophage/microglia infiltration and activation in hippocampus and basal ganglia of HAART-treated patients as compared to samples from the time before HAART [6]. This is in line with more recent neuropathological descriptions reporting various forms with severe HIVE and white matter injury, extensive perivascular lymphocytic infiltration, 'burnt-out' forms of HIVE and aging-associated amyloid accumulation with Alzheimer's-like neuropathology [30].
Earlier histological studies in specimens from HIV-1 infected humans found that lymphocytes and monocytes enter the brain [108]. The pathophysiological relevance of CNS invading lymphocytes in HAD is still not clearly established [7], while in contrast an increased number of microglia and macrophages correlates with severity of pre mortem HAD [6, 41, 108]. However, infiltrating lymphocytes and activated microglia in brains with HIV-1 encephalitis might show strong immunoreactivity for IL-16, a natural ligand of CD4 that inhibits HIV-1 propagation. Thus, lymphocytes might contribute to an innate antiviral immune response in the CNS in addition to microglia [137]. Furthermore, different lymphocyte populations produce IL-4 and interferon (IFN)-y, two cytokines with important roles in immune regulation that seem to induce in microglia a cytoprotective phenotype [15]. In contrast, lipopolysaccharide (LPS) and aggregated beta-amyloid appear to induce a cytotoxic microglial phenotype, perhaps comparable to what HIV-1 or its protein components, such as gp120, achieve.
The blood-brain-barrier (BBB) also plays an important role in HIV infection of the CNS [36, 102]. Astrocytes and microglia produce chemokines - cell migration/chemotaxis inducing cytokines - such as monocyte chemoattractant protein (MCP)-1, which appear to attract peripheral blood mononuclear cells across the BBB into the brain parenchyma [7]. In fact, an increased risk of HAD has recently been connected to a mutant MCP-1 allele that causes increased infiltration of mononuclear phagocytes into tissues [44]. As an alternative to HIV entry via infected macrophages, it has been suggested that the inflammatory cytokine TNF-a, promotes a paracellular route for the virus across the BBB [32].
Altogether, it seems that a vicious cycle of immune dysregulation, inflammation and BBB dysfunction is required on the side of the host to allow sufficient entry of infected or activated immune cells into the brain and to permit neuronal injury [5, 43, 58, 60, 66, 96, 131]. Variations of the viral proteins gp120 and Tat on the part of the virus might also influence the timing and extent of events allowing viral entry into the CNS and subsequent neuronal injury [98, 113].
The neuropathology observed in post mortem specimens from HAD patients in combination with multiple studies using both in vitro and animal models of HIV-induced neurodegeneration have all contributed to a fairly complex model for the pathomechanism of HAD. The available information strongly suggests that the pathogenesis of HAD might be most effectively explained when viewed as similar to the multi-hit model of oncogenesis. Fig. 1 shows a model of potential intercellular interactions and alterations of normal cell functions that can lead to neuronal injury and death while Fig. 2 depicts potentially impaired neuronal renewal mechanisms in the setting of HIV infection [58, 60]. Macrophages and microglia can be infected by HIV-1, but they can also be stimulated by factors released from infected cells. These factors include cytokines and shed viral proteins such as gp120. Variations of the HIV-1 envelope protein gp120, in particular in its V1, V2 and V3 loop sequences, have been implicated in modulating the neurotoxicity of macrophages and microglia [113]. Factors secreted by activated microglia affect all cell types in the CNS, resulting in upregulation of cytokines, chemokines and endothelial adhesion molecules [36, 43, 58, 60, 66]. Some of these factors may directly or indirectly contribute to neuronal damage and apoptosis. Directly neurotoxic factors released from activated microglia and macrophages include excitatory amino acids (EAAs) and related substances, such as quinolinate, cysteine and a not completely characterized amine compound named 'Ntox' [39, 43, 50, 58, 60, 66, 78, 135]. EAAs can trigger neuronal apoptosis through a process known as excitotoxicity. This detrimental process involves excessive Ca2+ influx and free radical (nitric oxide and superoxide anion) formation by overstimulation of glutamate receptors [78], activation of stress-associated protein kinases and caspases and production of proinflammatory lipids [43, 56, 58, 66, 86, 100]. Certain HIV proteins, such as gp120, Tat and Vpr, may also exert some direct neurotoxicity, in particular when neurons are cultured in isolation or separated from glial cells prior to exposure in order to detect these direct effects [80, 86, 91, 92]. It is important to note that toxic viral proteins among factors released from microglia and glutamate set free by astrocytes may all act in concert to provoke neuroinflammation and degeneration, even in the absence of extensive viral invasion of the brain. Furthermore, HAD might share the
critical involvement of neuroinflammation and microglial activation with several other neurodegenerative diseases, such as Alzheimer's disease, Multiple Sclerosis, Parkinson's disease and Frontotemporal Lobe Dementia [11].
CHEMOKINES AND THEIR RECEPTORS IN HIV-1 INFECTION AND NEURONAL DEATH
Chemokine receptors belong in the large category of seven transmembrane-spanning domain proteins. As Gprotein coupled receptors, they can trigger a wide range of intracellular signals [81]. Originally, chemokines and their receptors were found to mediate leukocyte trafficking and to contribute intimately to the organization of inflammatory responses of the immune system, but they are now recognized as contributors to many more physiological and pathological processes [25, 81, 103, 123]. The additional functions include the intricate control of organogenesis, including hematopoiesis, angiogenesis, and development of heart and brain [81, 83, 118, 139]. Moreover, chemokines and their receptors are indispensable for maintenance, maturation and migration of hematopoietic and neural stem cells [71, 123]. However, to date the most prominent pathological function of certain chemokine receptors seems to be the mediation of HIV-1 infection [25, 81, 103].
Infection of macrophages and lymphocytes by HIV-1, systemically and in the brain, can occur after binding of the viral envelope protein gp120 to one of several possible chemokine receptors in conjunction with CD4. Depending
on the exact type of gp120, different HIV-1 strains may use CCR5 (CD195) and CCR3, or CXCR4 (CD184), or a combination of these chemokine receptors to enter target cells [25, 48, 103]. Generally, T-cells are infected by 'Ttropic' viruses via the a-chemokine receptor CXCR4 and/or the a-chemokine receptor CCR5. In contrast, macrophages and microglia are infected by 'M-tropic' HIV-1 primarily via
CCR5 and CCR3, but the a-chemokine receptor CXCR4 may also be involved [17, 48, 94, 104]. Indeed, usage of CCR5 is neither necessary nor sufficient for macrophage tropism [45, 75]. However, the crucial role of chemokine receptors on several levels in HIV-1 disease has become increasingly obvious in recent years [127]. Usually CCR5- preferring HIV-1 strains (R5-tropic) are transmitted between humans, and individuals lacking CCR5 are highly resistant to primary HIV infection [79]. CXCR4-using viruses (X4- tropic) occur in about 50 % of infected individuals later in the course of HIV-1 disease and indicate progression to AIDS [94]. However, in many HIV-1/AIDS patients the switch in coreceptor usage does not occur and R5-tropic viruses evolve over time into more cytopathic variants possessing higher CCR5 affinity combined with reduced CD4 dependence [45]. X4-tropic HIV-1 strains might be inhibited in mucosal transmission due to the high expression of stromal cell-derived factor-1 (SDF-1, CXCL12), the natural ligand for CXCR4, in genital and rectal epithelium [4]. On the other hand, the CCR5 ligands MIP-1a (CCL-3), MIP-1beta (CCL-4) and RANTES (CCL-5) are prominently produced by Tlymphocytes and suppress CCR5-mediated HIV-1 infection [20]. In fact, individuals exposed to HIV-1 who remain yet uninfected or become infected but remain long-term asymptomatic and do not progress to AIDS have been found to express high levels of the same CCR5-binding beta-chemokines [106].
Neurons and astrocytes express among other chemokine receptors also CCR5 and CXCR4 [7, 95], although these cells are not thought to harbor productive HIV-1 infection under in vivo conditions. However, several in vitro studies strongly suggest that CXCR4 is prominently involved in HIV-associated neuronal damage while CCR5 may additionally serve a protective role [49, 59, 91, 92].
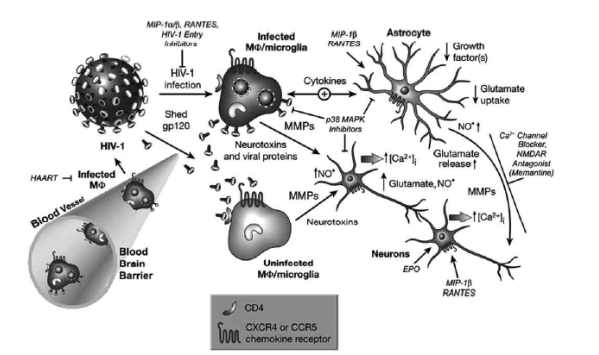
Fig. (1). Current model of HIV-1 neuropathology indicating presumably toxic or protective factors and potential sites for therapeutic intervention (protective factors are shown in italic). Neuronal injury and death induced by HIV-1 infection: Immune-activated and HIV-infected, brain-infiltrating macrophages (M/F) and microglia release potentially neurotoxic substances. These substances include quinolinic acid and other excitatory amino acids such as glutamate and L-cysteine, arachidonic acid, PAF, NTox, free radicals, TNF-a, and probably others. These factors from MF/microglia and also possibly from reactive astrocytes contribute to neuronal injury, dendritic and synaptic damage, and apoptosis as well as to astrocytosis. Entry of HIV-1 into MF/microglia occurs via gp120 binding, and therefore it is not surprising that gp120 (or a fragment thereof) is capable of activating uninfected MF/microglia to release similar factors to those secreted in response to productive HIV infection. MF/microglia express CCR5 and CXCR4 chemokine receptors on their surface in addition to CD4 and viral gp120 binds via these receptors. Some populations of neurons and astrocytes have been reported to also possess CXCR4 and CCR5 receptors on their surface, raising the possibility of direct interaction with gp120. MF/microglia and astrocytes have mutual feed-back loops (bidirectional arrow). Cytokines participate in this multi-cellular network in several ways. For example, HIV infection or gp120-stimulation of MF/microglia enhances their production of TNF-a and IL-1é (cytokines-arrow). The TNF-a and IL-1é produced by MF/microglia stimulate
astrocytosis. Arachidonate released from MF/microglia impairs astrocyte clearing of the neurotransmitter glutamate and thus contributes to excitotoxicity. In conjunction with cytokines, the a-chemokine SDF-1 stimulates reactive astrocytes to release glutamate in addition to the free radical nitric oxide [NO�], which in turn may react with superoxide (O2�-) to form the neurotoxic molecule peroxynitrite (ONOO-). NO might also activate extracellular matrix metalloproteinases (MMPs), which can then proteolytically affect neurons, and also cleave membrane-anchored fractalkine [60]. Neuronal injury is primarily mediated by overactivation of NMDARs with resultant excessive influx of Ca2+. This, in turn, leads to overactivation of a variety of potentially harmful signaling systems, the formation of free radicals, and release of additional neurotransmitter glutamate. Glutamate subsequently overstimulates NMDARs on neighboring neurons, resulting in further injury. This final common pathway of neurotoxic action can be blocked by NMDAR antagonists. For certain neurons, depending on their exact repertoire of ionic channels, this form of damage can also be ameliorated to some degree by calcium channel antagonists or non-NMDAR antagonists. Additionally, MIP-1beta and RANTES, agonists of beta chemokine receptors, which are present in the CNS on neurons, astrocytes and microglia, can confer partial protection against neuronal apoptosis induced by HIV/gp120 or NMDA.
Intact HIV-1, as well as picomolar concentrations of viral envelope gp120, can induce neuronal death via CXCR4 and CCR5 receptors in cerebrocortical and hippocampal neurons and neuronal cell lines from humans and rodents [17, 35, 49, 53, 59, 91, 104, 128]. Recently, we have further investigated the role of chemokine receptors in the neurotoxicity of gp120 using mixed neuronal/glial cerebrocortical cultures from rat and mouse. We found that gp120 from CXCR4
(X4)-preferring as well as CCR5 (R5)-preferring and dual tropic HIV-1 strains all were able to trigger neuronal death. While gp120 from one out of two X4-preferring HIV-1 strains showed no longer neurotoxicity in CXCR4-deficient
cerebrocortical cultures, dual tropic gp120SF2 exerted surprisingly even greater neurotoxicity in CCR5 knockout cultures compared to wild-type or CXCR4-deficient cerebrocortical cells [60]. These findings are consistent with a primarily neurotoxic activation of CXCR4 by gp120. In contrast, CCR5 might at least in part stimulate cytoprotective signals depending on the HIV-1 strain from which a given envelope protein originated. Furthermore, we observed earlier that the
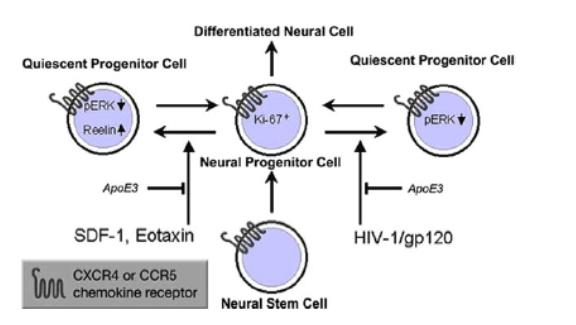
Fig. (2). Current model of HIV-1 interference with the function of neural progenitor cells and potential sites for therapeutic intervention (protective factors are shown in italic): Exposure to chemokines, SDF-1 and Eotaxin, or HIV 1/gp120 of mouse or human neural progenitor cells (NPCs) reduces proliferation and promotes quiescence. ApoE3 inhibits these effects on NPCs. NPCs express nestin and show decreased proliferation as judged by decreased BrdU incorporation. However, NPCs do not undergo apoptosis, as evidenced by lack of TUNEL staining and nuclear condensation under the same conditions ([67, 68] and S. Okamoto, S. McKercher, M. Kaul, and S.A. Lipton, unpublished). Modified from [58, 60].
CCR5 ligands MIP-1beta and RANTES protect neurons against gp120-induced toxicity [59].
In mixed neuronal/glial cerebrocortical cultures that mimic the cellular composition of the intact brain, HIV/gp120-induced apoptotic death appears to be mediated predominantly via the release of microglial toxins rather than by direct neuronal damage [12, 17, 35, 39, 53, 59, 128]. In fact, both the absence or inactivation of macrophages and microglia basically abrogates the neurotoxicity of HIV/gp120 in mixed neuronal/glial cultures [35, 39, 40, 59]. On the other hand, nanomolar concentrations of SDF-1a/beta interacting with CXCR4 can induce apoptotic death of neurons independent of microglial activation, suggesting a
possible direct interaction with neurons and astrocytes [9, 59, 138]. In contrast to these findings, it has been reported that SDF-1a also can provide neuroprotection from X4-preferring gp120-induced damage of isolated hippocampal or cortical neurons and mixed glial/neuronal cerebrocortical cells [64, 91, 92]. While the reasons for the apparently contradictory findings remain to be elucidated, differences in the preparation and age of the respective cell culture models might provide one possible explanation. When SDF-1 was
neuroprotective, it was found to activate Akt (protein kinase B) and mitogen-activated protein kinases (MAPKs) [63] and regulate the expression and localization of cell cycle proteins [62, 64].
Since at least in vitro inhibition of microglial activation suffices to prevent neuronal death after gp120 exposure, it seems likely that stimulation of CXCR4 (or CCR5) in macrophages / microglia is a prerequisite for the neurotoxicity
of gp120 [59, 104]. In contrast, SDF-1 might directly activate CXCR4 in astrocytes and neurons to trigger neuronal death, for example, by reversing glutamate uptake in astrocytes [9, 49, 58, 59]. SDF-1 is synthesized by astrocytes, macrophages, neurons and Schwann cells [89, 138]. Elevated levels of SDF-1 mRNA have been detected in HIVE as compared to uninfected controls [7, 138], and protein expression of SDF-1 also appears to be elevated in the brains of HIV patients [69]. To what degree the increased expression of SDF-1 aggravates neuronal damage by HIV-1 remains to be shown. In fact, we had previously observed in vitro that intact SDF-1 can be toxic to mature neurons in a
CXCR4-dependent manner [59, 60, 138]. Additionally, it was recently discovered that cleavage of SDF-1 by matrix metalloproteinases (MMPs) may contribute to neuronal injury and thus HAD via a likely non-CXCR4-mediated mechanism [136]. Importantly, increased expression and activation of MMPs, including MMP-2 and MMP-9, were detected in HIV-infected macrophages and also in post mortem brain specimens from AIDS patients compared with uninfected controls [55]. As elegantly demonstrated by Power and colleagues, MMP-2 released from HIV-infected macrophages is able to proteolytically remove four amino acids from the N-terminus of SDF-1. The truncated a-chemokine is an even more powerful neurotoxin than fulllength SDF-1, but it seems no longer to bind CXCR4 [136].
Neuropathological features that are similar to the findings in brains of AIDS patients, such as reactive astrocytosis, increased number and activation of microglia, reduction of synapto-dendritic complexity, loss of large pyramidal neurons and even induction of MMP-2 all occur in the CNS of transgenic (tg) mice expressing HIV-1/gp120 [84, 122]. These gp120 tg mice develop significant behavioral deficits, such as extended escape latency, and reduced swimming velocity and spatial retention [22]. These findings suggest that the HIV-1 surface glycoprotein and engagement of the host's viral envelope receptors may well be sufficient to initiate neuronal injury and behavioral alterations.
HIV-1/GP120 AND CHEMOKINES AFFECT NEURAL STEM AND PROGENITOR CELLS
The CXCR4-SDF-1 receptor-ligand axis plays an important role in the physiological function of hematopoietic and neural stem cells [7, 123]. This fact indicates a potential of HIV-1 and its envelope protein to directly interfere with biological functions of neural stem and progenitor cells.
In cultures of primary mouse and human neural progenitor cells obtained from fetal tissue, cells stain positively for the neural stem cell marker nestin and readily undergo cell division. After several rounds of proliferation, the progenitors
exit the cell cycle and express neuronal markers such as betaIII-tubulin (TuJ1). Our immunocytochemical studies showed that the progenitors expressed CXCR4 and CCR5. Treatment with HIV-1/gp120 reduced the number of progenitors and differentiating neurons. Accounting for these observations, we found that gp120 inhibited proliferation of neural progenitor cells without producing apoptosis.
The
resulting decrease in neural stem cell proliferation engendered by gp120 also meant that there were fewer progenitor cells present to differentiate in neurons, thus impairing neurogenesis (S. Okamoto, S. McKercher, M. Kaul, and S.A. Lipton, unpublished). These findings were complemented and extended by others using commercially-generated human neural progenitor cells [67, 68]. In those experiments, chemokines promoted the quiescence and survival of human neural progenitor cells via stimulation of CXCR4 and CCR3 and a mechanism that involves downregulation of extracellularly regulated kinase-1 and -2 (ERK-1/2) with simultaneous upregulation of the neuronal glycoprotein reelin [67]. Exposure to HIV-1 caused quiescence of neural progenitors, again through engagement of CXCR4 and CCR3. The coat protein HIV-1/gp120 reportedly downregulated ERK-1/2 but had no effect on Reelin [68]. Interestingly, the effects of both the chemokines and HIV-1/gp120 were reversible and could be inhibited with recombinant Apolipoprotein E3 (ApoE3), but not ApoE4. Although it is widely accepted that HIV-1 fails to productively infect neurons, it has been reported that neural progenitor cells are permissive to the virus [86]. The apparent ability of HIV-1/gp120 to interfere with the normal function of neural progenitor cells suggested the possibility that HAD might develop as a consequence not only of injury and death of existing neurons but also due to virus-induced disturbance of potential repair mechanisms in the CNS (Fig. 2).
NEURONAL INJURY AND DEATH IN HIV-1 INFECTION AND HAD
Despite continuing progress in uncovering the pathologic processes, it remains a controversial topic how exactly HIV-1 infection provokes neuronal injury and death as well as neurocognitive and motor impairment [43, 60, 66, 86]. While
there is general agreement that HIV does not infect neurons, the mechanism of neuronal damage remains in question. There is evidence for neuronal injury by various viral proteins; including Tat, Nef, Vpr and the Env proteins gp120
and gp41 [2, 12, 58, 60, 65, 86, 99, 111]. These findings have led to at least two different hypotheses on how HIV-1 initiates neuronal damage in the brain. The hypotheses can be described as the "direct injury" hypothesis and the "indirect' or "bystander effect" hypothesis. These two hypotheses are not mutually exclusive, and while the available data support a role for both, an indirect form of neurotoxicity seems to predominate in a setting where glial and neuronal cells are present [36, 43, 58, 60, 86].
The hypothesis that HIV proteins can directly injure neurons without requiring the intermediary function of nonneuronal cells (microglia and/or astrocytes) is supported by experiments showing that viral envelope proteins are toxic in serum free primary neuronal cultures [91, 92] and in neuroblastoma cell lines [49]. The impact of neurotoxic cytokines and EAAs secreted from non-neuronal cells is minimized in these experimental paradigms because serum-free neuronal cultures contain few if any non-neuronal cells, and neuroblastoma lines do not contain cells of other phenotypes. The HIV envelope protein gp120 interacts with several members of the chemokine receptor family (see above), and the direct form of HIV-induced neuronal injury may be mediated by chemokine receptor signaling in the absence of CD4 [49]. Indeed, experiments aimed at blocking chemokine receptor signaling can in some cases prevent HIV/gp120-induced neuronal apoptosis [59, 91, 92, 138]. In another study, gp120 has been reported to interact at nanomolar concentrations with the glycine binding site of the N-methyl-D-aspartatetype glutamate receptor (NMDAR) [33], suggesting an alternative mechanism by which HIV/gp120 may directly affect neuronal viability. The HIV-protein Tat (HIV/Tat) can be taken up into PC12 cells by a receptor mediated mechanism [80] and may also have a direct effect on neurons by potentiating the response to excitotoxic stimuli (reviewed in [86]). Experiments using cultured hippocampal neurons revealed that the HIV-protein Vpr may be directly neurotoxic through formation of a cation permeable channel [111]. Of note, the interpretation of most of these in vitro findings must take into account the fact that the experimental results were obtained in the absence of non-neuronal cells and therefore a predominantly indirect effect would not be detected. Although the absence of non-neuronal cells allows the study of potential direct effects of viral proteins on neurons, the
pathophysiological relevance of these results remains uncertain because neurons in the brain never encounter the potential toxins in the absence of glial cells.
In brains from HAD patients apoptotic neurons do not co-localize with infected microglia [3, 104], supporting the hypothesis that HIV infection causes neurodegeneration through the release of soluble factors. Therefore, the propensity for cell-cell interactions mandates that disease pathogenesis in vitro be approached in a 'mixed' neuronal/glial primary culture system that resembles the type and proportion of cells usually found in the intact brain (Fig. 1). Systems designed to study the effect of soluble factors released from microglia have included mixed cerebrocortical cultures from human fetal brain directly infected with HIV [104], severe combined immunodeficiency (SCID) mice inoculated with HIV-infected human monocytes [134], gp120 transgenic mice [121], and mixed human and rodent cerebrocortical cultures exposed to picomolar concentrations of the envelope protein HIV/gp120 [12, 26, 40, 53, 59, 93, 128].
The pronounced response of non-neuronal cells to HIV infection or shed HIV proteins in such in vitro and in vivo models provided evidence for a predominantly indirect neurotoxic effect. Much of the data supporting the hypothesis of indirect neuronal injury stems from experiments designed to
examine the toxicity of HIV envelope proteins or supernatants of infected macrophages [12, 26, 39, 40]. Picomolar concentrations of HIV/gp120 induce injury and apoptosis in primary rodent and human neurons [12, 26, 59, 104, 138]. In our hands, the predominant mode of HIV/gp120 neurotoxicity to cerebrocortical neurons requires the presence of macrophages/microglia [39, 58, 59]. Indeed, HIV-1 infected or gp120 stimulated mononuclear phagocytes release neurotoxins that stimulate the NMDAR, as described earlier. NMDAR antagonists can ameliorate neuronal cell death in vitro due to HIV-infected macrophages or purified recombinant gp120 [17, 26], and in vivo in gp120 transgenic mice [121].
Activation of ionotropic glutamate receptors in neurons initiates under physiological conditions a transient depolarization and excitation. While AMPARs mediate a fast component of excitatory postsynaptic potentials, NMDARs underlie a slower component. Under resting conditions, Mg2+ blocks the NMDAR-associated ion channel. Presynaptic release of glutamate and consequent depolarization of the postsynaptic neuronal membrane via AMPAR-coupled channels relieves the Mg2+ block, and allows subsequent controlled Ca2+ influx through the NMDAR-coupled ion channel. This voltage-dependent regulation of NMDAR function results in activity-driven synaptic modulation [10, 24]. However, extended and/or excessive NMDAR activation and consequent excitotoxicity is triggered by sustained elevation of the intracellular Ca2+ concentration, compromised mitochondrial function and cellular energy metabolism, and resultant free radical formation [24, 77, 105].
These NMDAR-mediated detrimental intracellular signals contribute to neuronal cell injury and subsequent death by apoptosis or necrosis, depending on the intensity of the initial insult [100]. If the initial excitotoxic insult is fulminant, for example, in the ischemic core of a stroke, the cells die early from loss of ionic homeostasis, resulting in acute swelling and lysis (necrosis). If the insult is more mild, as seen in several neurodegenerative disorders including HAD, neurons enter a delayed death pathway known as apoptosis [100, 109]. Neuronal apoptosis after excitotoxic insult involves Ca2+ overload, activation of p38 MAPK and p53, release of cytochrome c and other molecules such as apoptosis- inducing factor (AIF) from mitochondria, activation of caspases, free radical formation, lipid peroxidation, and chromatin condensation [7, 34, 35, 120]. In line with these observations is a report that in human neurons, CXCR4 mediates the toxic effect of gp120 via a process involving the synthesis of ceramide and NADPH-dependent production of superoxide radicals [54]. Activated caspase-3 and p53 are prominently detected in neurons of brains from HAD patients [34]. Furthermore, in vitro, neuronal caspases-3, -8 and -9 are involved and p53 is indispensable in neurons (and microglia) for HIV-1/gp120 to cause neurotoxicity [34, 35]. It has been suggested that CXCR4 and p53 are connected through signaling pathways that mediate toxic or protective mechanisms depending on whether gp120 or SDF-1 acts as the ligand [64]. In contrast to gp120IIIB, SDF-1 was found to have a neuroprotective effect. It activated Akt and MAPKs [63] and regulated the expression and localization of cell cycle proteins [62, 64]. SDF-1 increased acetylation of p53 and p21 as well as the expression of retinoblastoma protein (Rb) while reducing the amount of phosphorylated Rb in the nucleus. Together with a reduction of the activity of the transcription factor E2F1, an overall anti-apoptotic effect was observed. In contrast, envelope protein of HIV-1IIIB caused the opposite effect to SDF-1 in the nucleus, triggered activation of Apaf-1 and promoted cell death. Besides these in vitro findings, changes from the normal expression pattern have been observed for the same cell cycle proteins mentioned above in post mortem brains derived from non-human primates with SIV encephalitis and humans with HIV encephalitis [57]. Interestingly, these changes to cell cycle proteins correlated with the presence of activated microglia and macrophages.
Excessive intracellular Ca2+ overstimulates nNOS and protein kinase cascades with consequent generation of deleterious levels of free radicals, including reactive oxygen species (ROS) and nitric oxide (NO) [100]. NO can react with ROS to form cytotoxic peroxynitrite (ONOO-) [100]. Oxidative processes and cellular distress are also reflected by alterations to the cellular lipid metabolism, and an increase in ceramide, sphingomyelin and hydroxynonenal has been implicated in the neurotoxic pathways associated with HAD [86].
In addition to the intracellular effects of NO and oxidative stress, we have recently identified an extracellular proteolytic pathway to neuronal injury mediated by these effectors. In this pathway, S-nitrosylation (transfer of NO to a critical cysteine thiol group) and subsequent oxidation serve to activate MMP-9 and possibly other MMPs [47]. Proteolytically active MMP-9 induces and promotes neuronal death presumably by disrupting the cellular mechanism(s) that allow essential attachment to the extracellular matrix and neighboring neurons.
In addition to chemokines, MMPs and EAAs, HIV-infected or gp120-activated microglia also release inflammatory cytokines, including TNF-a and IL-1beta [107, 130]. Interestingly, TNF-a can under certain conditions protect neurons by stabilizing intracellular Ca2+ homeostasis [18]. However, among other actions, both TNF-aand IL-1beta can stimulate release of L-cysteine from macrophages, and pharmacologic blockade of IL-1beta or antibody neutralization of TNF-a prevents this release [135]. Under physiological or pathophysiological conditions, L -cysteine can stimulate NMDARs and lead to neuronal apoptosis [135]. TNF-a is capable of stimulating apoptosis in human neurons [119], but an indirect route of injury cannot be excluded, such as autoor paracrine inflammatory stimulation of macrophages and microglia to produce neurotoxins. Expression of TNF-a and
its receptor are elevated in brains from patients with HAD [130]. TNF-a has direct effects on glutamate neurotransmission by increasing the synaptic expression of AMPARs and inhibiting long-term potentiation (LTP) in a p38 MAPK-dependent
manner [14, 110]. TNF-a can promote neurotoxicity by facilitating glutamate excitotoxicity through inhibition of astroglial glutamate transporters [110] or even provoking neurotransmitter release from glial cells [8, 9]. Experiments aimed at addressing the question of interactions between neurotoxins associated with HAD have revealed that TNF-a and HIV/Tat synergize to promote neuronal death, and this effect is prevented by antioxidants [104]. It remains possible that TNF-a can activate caspases within neurons via TNF-a receptor-1 (TNFR1), since TNFR1 is found on at least some neurons, and it can trigger caspase-8 activation. Indeed, we have found that antibody neutralization of TNF-a or inhibition of caspase-8 prevents the neurotoxicity of HIV/gp120 in cultured cerebrocortical neurons [34]; and caspase-8 activity can directly or indirectly activate caspase-3, leading to apoptosis. Another member of this enzyme
family, caspase-1, converts inactive pro-IL-1beta into mature, active IL-1beta, thus generating a factor with a complex role in potential promotion and limitation of neuroinflammation and neuronal injury [5]. These findings suggest that inflammatory cytokines, including TNF-aand IL-1beta, may have important regulatory roles in HIV-associated neuropathology [5, 43, 58, 60, 66, 119, 135].
TNF-related apoptosis-inducing ligand (TRAIL) [37, 117] is a type II integral membrane protein and a member of the TNF superfamily, thus is closely related to FAS ligand [133]. TRAIL interacts with at least five unique receptors found on a variety of cell types. TRAIL-expressing macrophages have been found in close proximity to caspase-3 positive neurons in brain tissue from HIV-1 patients with encephalitis [116]. In addition, in a model combining NODSCID mice and HIV-infected human peripheral blood mononuclear cells, it was shown recently that addition of lipopolysaccharide (LPS) causes the infected human cells to infiltrate the murine brain and to cause neuronal apoptosis.
This effect was not only specific for macrophage-tropic HIV-1 but also prevented by a neutralizing anti-TRAIL antibody [97]. These findings strongly suggested a role for TRAIL in the induction of neuronal death by infected human macrophages. However, even though TRAIL has been reported to induce apoptosis in brain cells [101], it remains to be shown, whether or not killing of neurons occurs as a consequence of a direct or indirect interaction.
Transgenic (tg) mice expressing HIV-1/gp120 in their CNS manifest neuropathological features that are similar to the findings in brains of AIDS patients, including reactive astrocytosis, increased number and activation of microglia, reduction of synapto-dendritic complexity, loss of large pyramidal neurons [122], and induction of MMP-2 [84]. In these tg mice, neuronal damage is ameliorated by the NMDAR antagonist memantine [121]. Memantine-treated gp120 tg and non-tg control mice maintain a density of presynaptic terminals and dendrites that is similar to untreated non-tg/wildtype controls but significantly higher than in untreated gp120 tg animals [121]. This finding confirms the hypothesis that the HIV-1 surface glycoprotein is sufficient to initiate excitotoxic neuronal injury and death. It also shows that an antagonist of NMDAR overstimulation can ameliorate HIV/gp120-associated neuronal damage in vivo [26, 121].
APPROACHES TO TREATMENT OR PREVENTION OF HIV-1 INDUCED NEURONAL DYSFUNCTION AND HAD
While HAART has tremendously improved the treatment of HIV-1 infection and disease in the periphery, an effective pharmacotherapy for HAD is still not available. However, even the most effective HAART protocol has not been able to eradicate HIV-1 from an once infected organism, because the virus hides in some infected cells and remains latent [19]. However, a recent proof-of-concept study has shown that valproic acid, a histone deacetylase 1 (HDAC1) inhibitor and a drug used to treat epilepsy, reduces the pool of latent virus [73]. It is still too early to assess if that approach will eventually allow to clear the brain of HIV-1, but it indicates that the possibility might exist in the future.
Previous approaches to cope with HAD reflect the challenging complexity inherent in the treatment of patients with AIDS (reviewed by Melton et al. [90] and [124]). Previous and current therapeutic approaches include various antiretroviral compounds, alone or in combination, such as Zidovudine, Didanosine, Zalcitabine, and Stavudine. Of these only Zidovudine has been found to cross the blood-brainbarrier to a certain extent. Although Zidovudine has a beneficial effect on HAD, the effect is not lasting. The other currently available anti-retroviral drugs may not penetrate the brain sufficiently to control the virus in the CNS. Thus, at the moment an efficient adjunctive treatment besides antiretroviral drugs is still needed.
Considering the evolving picture of HAD pathogenesis described above, several potential therapeutic strategies toattenuate neuronal damage are worth exploring. Among others, agents warranting consideration include NMDAR blockers, cytokines, chemokines, chemokine and cytokine receptor antagonists, p38 MAPK inhibitors, caspase inhibitors, and antioxidants (free radical scavengers or other inhibitors of excessive nitric oxide or reactive oxygen species).
Drugs aiming at prevention or limitation of excitotoxic neuronal insult, such as NMDAR antagonists, have been shown to attenuate neuronal damage due to either HIV-infected macrophages or HIV/gp120, both in vitro and in vivo. The open-channel blocker, memantine, prevents excessive NMDAR activity while largely sparing physiological function [76]. Also, unlike other NMDAR antagonists tested in clinical trials to date, memantine has proven both safe and effective in a number of phase III clinical trials for Alzheimer's disease and vascular dementia. The outcome of a large, multi-center NIH-sponsored clinical trial testing this agent in patients with HAD has suggested some benefit, and improved second generation drugs are currently under development. Previous, small clinical trials of a voltageactivated calcium channel blocker, nimodipine, and a PAF inhibitor suggested some therapeutic benefit but were not conclusive [124]. A separate clinical trial with the antioxidant drug selegiline is aimed at combatting the effects of excitotoxicity by minimizing the impact of free radicals [124].
Mood changes approaching the extent of disorders are one of many problems associated with HIV-1 disease. Thus, Lithium has been suggested as a treatment for HAD because it also affects the cytoprotective phosphoinositol-3 kinase (PI3K)/Akt (protein kinase B)/GSK-3beta pathway [29].
Recently, we have reported that the cytokine erythropoietin (EPO) may not only be effective in treating anemia but also for protecting neurons against inflammatory and excitotoxic injury, because it prevents NMDAR-mediated and HIV-1/gp120-induced neuronal death in mixed cerebrocortical cultures [23]. Since EPO is already clinically approved for the treatment of anemia, human trials of EPO as a neuroprotectant from HIV-associated dementia may be expedited [23].
Chemokine receptors allow HIV-1 to enter cells and as such are major potential therapeutic targets in the fight against HIV-1 infection and AIDS [94]. Inhibitors of
CXCR4 and CCR5 prevent HIV-1 entry and are being assessed in clinical trials [94]. However, the benefit of chemokine receptor antagonists for HIV-associated neurological complications, although likely, remains to be shown [36, 58, 60]. Interestingly, as alluded to above, HIV-infected patients with higher CSF concentrations of the beta -chemokines MIP-1a/beta and RANTES performed better on neuropsychological measures then those with low or undetectable
levels [74]. Additionally, certain chemokines have been experimentally
shown to protect neurons, even though the virus does not productively infect neurons. In particular, beta-chemokines (acting on CCR5 receptors) and fractalkine prevent gp120-induced neuronal apoptosis in vitro [13, 59, 92], and, similarly, some beta-chemokines can ameliorate NMDAR-mediated neurotoxicity [13]. All these findings support the hypothesis that selected beta-chemokines may represent a potential treatment modality for HAD.
Given the substantial impact of inflammatory diseases on health in general, the pharmaceutical industry is currently developing p38 inhibitors for a variety of inflammatory- and stress-related conditions, such as arthritis, and this may expedite trials for CNS indications including HAD. The rationale is that p38 MAPK inhibitors have been shown to reduce or abrogate neuronal apoptosis due to excitotoxicity, HIV/gp120 exposure, or ß-chemokine (SDF-1) toxicity [59].
Neuronal death by apoptosis appears to be one of the hallmarks of neurodegenerative diseases including HAD [3]. Caspases carry out the apoptotic program and, as detailed above, they have been implicated in HIV-related neuronal damage [34]. Therefore, caspase inhibitors may be helpful in preventing detrimental neuronal loss. However, caspase inhibitors are not currently available in a form deliverable to the CNS or targeted to degenerating neurons. While further advances in the caspase field might eventually produce such drugs, care must be taken to avoid disturbance of physiologic turn-over of cells or even promote oncogenic processes.
Altogether, post mortem studies of human AIDS brains and experimental evidence regarding the pathologic mechanism of HAD indicate that synergy between inflammatory and excitatory pathways to neuronal injury and death may, at least in part, be shared with other CNS disorders including stroke, spinal cord injury and Alzheimer's disease. Newly developed therapeutic strategies for HAD will therefore likely benefit the treatment of several other neurodegenerative diseases and possibly vice-versa.
|
|
| |
| |
|
|
|