 |
|
|
| |
HIV drug resistance at IDSA/ICAAC
|
| |
| |
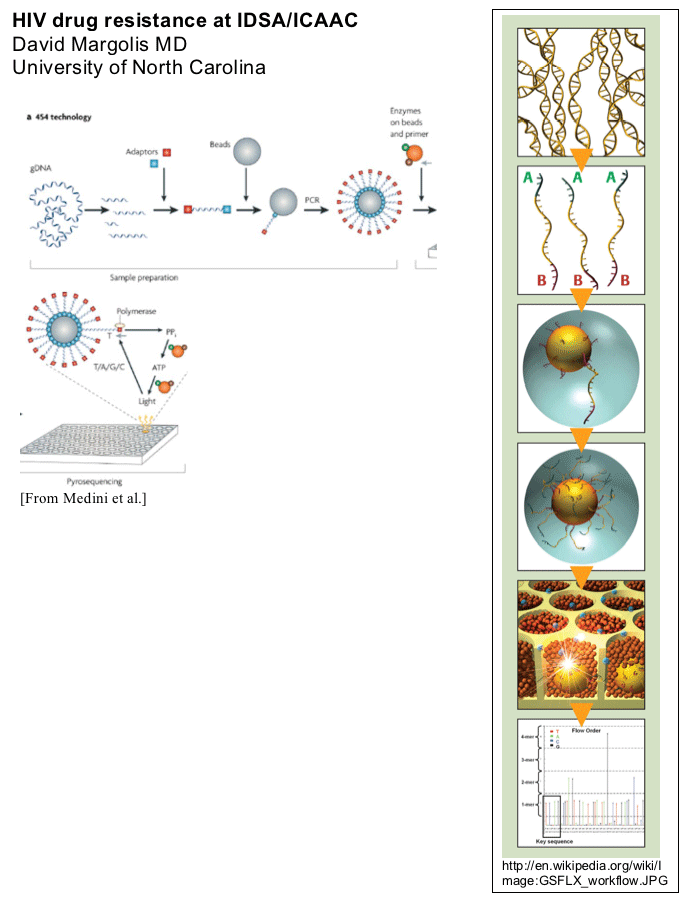
The depths of resistance: Ultra-deep sequencing is a new and mind-boggling technique that allows the massively parallel sequencing of thousands of individual DNA or RNA molecules. The 454 Technologies machine uses sequencing a two-step approach. First, the DNA is fragmented and small DNA adaptors attached. DNA sequences are then attached to a bead and the beads are PCR amplified within droplets of oil-water emulsion. This generates multiple copies of the same DNA sequence on each bead contained within an oil droplet. Next beads are poured over a plate fabricated with computer chip technology that has picolitre-sized wells that are only large enough to fit one bead. Pyroophosphate-based
sequencing is performed within these tiny wells on each DNA fragment. The addition of a nucleotide releases inorganic pyrophosphate (PPi), which generates ATP used by luciferase in the reaction mix to generate a light signal. This signal is read by a detector, which notes the addition of a bases in the glowing wells. The cycle is repeated for each of the four bases, and the 454 machine's hardware keeps track of which bases are being added in each of the machines wells. The average read length has already increased to approximately 250 bp, and future developments will probably increase allow sequencing of longer DNA fragments [Medini et al. Nature Reviews Microbiology 2008]
Rich D'Aquilla at Vanderbilt, with collaborators in Nashville and at GSK dove into the depths of 454 sequencing, and reported what they found in patients who were treated with ART that included tenofovir (TDF) or abacavir (ABC) [abstr. H-897]. They studied stored samples from 16 patients who initially received ART with good response, and then had viremia while on these NRTIs. In the group of 16, none had suffered more than 3 episodes of virological failure while receiving ART, and for 9 of these patients this was their first episode of virological failure.
HIV sequences recovered from plasma at more than one time point were analyzed between amino acids 56 and 120 of HIV reverse transcriptase, a span of sequence easily covered by 454 technology. Of note, this is only a partial sequence of HIV RT, and does not reach some areas of where important mutations are know to occur (eg. Q151M, L210W, T215F/Y, K219Q/E). This is likely due to the cost of sequencing long regions by ultra-deep sequencing, but does allow the examination of areas of important mutations that may differentially appear on ABC or TDF therapy. TDF and ABC are associated with K65R and L74V mutations, respectively. Other NRTI-associated mutations in this region are described (A62V, K65N, K70E/G, L74I, V745I/M/T, F77L) and Y115F is reported in associated with ABC resistance.
The investigators thought it was likely that clinical sequencing studies often failed to detect low levels of these important mutations during viremia on TDF or ABC. High-throughput sequencing by 454 technology generates errors at a calculated probability of less than 1 per 1000. A median of 39,508 sequences were analyzed at each time point.
In three patients failing TDF-containing ART, no mutations (< 0.01%) were detected. Three other patients did have mutations detected; these 3 patients had K65R in most or all (56-98%) of their individual circulating viral sequences, and other mutations such as D67G/N or L74V in in minority (2-18%) of the circulating virus. In two of these patients, later sequencing did not show very much change in the viral populations at these resistance-related sites.
Similarly, in six patients failing ABC-containing ART, no NRTI mutations (< 0.01%) were detected, although in one the NNRTI mutation K103N was found in 98% of the sequences. Three other patients did have mutations detected. Again, these were all K65R, but instead of comprising most or all of the circulating sequence population, only rare circulating viruses (0.87-1.73%) encoded K65R. Other minority populations with mutations (D67G/N or K70R) were found at frequencies of 0.81-6.57% in two of these 3 patients. In the third patient nearly all (97-99%) of the circulating virus encoded D67N and K70R. Of note, and somewhat surprisingly, the L74V mutation that might be expected in patients failing ABC was not seen in any of the 9 patients failing ABC.
One patient on zidovudine (AZT) was studied and not found to have mutations in the region of RT, but did have the NNRTI mutation K103N. In all the patients, no other NNRTI mutations were seen, and it was reported from the podium that no protease mutations were seen (presumably by standard sequencing).
So overall, most patients (6 or 9 on ABC and 3 of 6 on TDF) had no NRTI drug resistance mutations detected deep down to very low frequencies. K65R was seen in most of the circulating virus in half of the patients failing TDF, but most of the circulating virus swarm in most patients failing ABC did not have mutations.
This study is limited by the retrospective nature of the cohort, its small size, and the lack of measures of drug concentration or (for what it might be worth) of adherence. However, the results are unexpected. Sensitive assays such as 454 deep sequencing deserve further careful study. As these instruments cost in the range of $1 million each, they will remain research tools for the time being, but as costs drop this technology might someday add to clinical management for some patients.
Resistance to HIV integrase inhibitors:
Daria Hazuda and her group presented further developments in the evolving story of resistance to integrase inhibitors. In a podium session on Sunday [H-898] Hazuda presented new analysis derived from the BENCHMRK 1 and 2 studies, pivotal licensure studies that demonstrated the efficacy of raltegravir (RAL) in patients with multi-drug resistant HIV and advanced disease.
Of 105 patients in both studies who developed detectable viremia above 400 copies/ml, plasma samples from 64 patients had sequence changes in the integrase region detectable. 30% of these patients had one sequence change, 44% had 2 changes, and nearly 27% had >2 integrase sequence changes. In 51 of these 64 patients, a plasma sample at a later time point could be examined. By this later time, only 7% now had only one integrase mutation, but now 47% had >2 integrase sequence changes, demonstrating clearly the development of new integrase mutations over time with continued virological failure.
When originally presented at CROI '07 in LA, two primary RAL resistance pathways were known; now three primary "pathways" to RAL resistance have been well documented:
a) a mutation at N155, most often to N155H, although up to 6 changes can be seen
b) a mutation at Q148 to Q148H, K, or R, although 5 other changes can be seen
c) a new pathway with a mutation at Y143 to Y143C or R, although 3 other mutations can be seen
A fourth pathway at 92 was rarely seen in BENCHMRK patients. Of note, after the initial Q148 mutation occurs, it appears to be followed by a second mutation(s) at nearby residues. These next mutations confer a much higher level of RAL resistance, and appear to mitigate replication defects conferred by the Q148 mutation. And as usual, the appearance of two or three RAL mutations confers high levels of resistance, and a greater ability of the mutant virus to replicate in culture ("fitness").
Over time in samples from the BENCHMRK studies, a bias for mutations in the Q148H emerged. Although the Q148H/R/K and N155H mutations had similar effects on RAL susceptibility and replication capacity in culture experiments, secondary mutations that followed Q148 conferred greater levels of resistance in the context of Q148 than those that followed N155H. In patients, the viruses seems to evolve towards the Q148 pathway which allows the greatest RAL resistance and the highest levels of replication, and hence a bias towards a predominance of Q148 in patients failing RAL over time.
Overall, this scenario is very reminiscent of protease inhibitor resistance. Experienced HIV treaters may be quick to draw that parallel and assume that the maintenance of RAL mutations may confer a clinical benefit despite continued viremia. If that assumption is made the following points should be considered:
1. there is as yet no clinical evidence for this hypothesis
2. accumulation of integrase mutations while on failing integrase therapy could compromise response to future integrase inhibitors. Although such drugs are in development, they obviously will not be available in the near term.
Along this line, Witmer of the Merck Research Laboratories group presented laboratory data on cross-resistance between RAL, elvitegravir (EVG), and second-generation experimental compounds [poster H-1232]. One of the three signature mutations in integrase (N155H, Q148H/K/R, or Y143C/H/R) that confer RAL resistance in clinical trials was cloned into a wild-type proviral DNA. Viruses were produced in the laboratory and evaluated for drug susceptibility in a single cycle infectivity assay (since to the Monogram assay for drug resistance and replication capacity).
Single mutations conferred resistance to RAL, and RAL resistance was increased by relevant secondary mutations. 7 secondary mutations could increase resistance with N155H, 8 mutations were show to increase resistance with the Q148 pathway, and 5 for the Y143 pathway. There was considerable overlap in secondary mutations between the different pathways (again, reminiscent of protease inhibitor resistance). All of these resistance viruses had declines in replication capacity (in vitro), but to a variable extent.
Most RAL-resistant viruses displayed greater resistance to EVG. This culture-based observation does not say much about the comparative clinical efficacy of the two integrase inhibitors, but consistent with previously reported data suggests that there will be clinical cross-resistance between the two drugs.
In contrast, the second-generation integrase inhibitors MK-0536 and MK-2048 displayed less cross resistance, but in general the degree of resistance to those compounds was much lower than to either RAL or EVG. However, a combination of mutations in the Q148 pathways including Q148K, 138K, and 140A resulted in 1000-fold resistance to MK-0536.
Provocatively, Dr. Hazuda discussed experiments in viral culture systems showing a "post-antibiotic effect" of RAL. Integrase inhibitors can completely block viral production in culture when added as late as 8 to 10 hours after infection. This is due to two to properties of integrase inhibitors. First, they act after viral entry and reverse transcription. Second, Dr. Hazuda showed data from an in vitro test tube experiment, in which integrase inhibitor drug remained bound to the pre-integration complex (PIC) - the complex of cellular and viral proteins that coat a completely reverse-transcribed HIV DNA genome and escort the PIC into the nucleus for integration - for many hours. This functionally irreversible binding suggests that once inhibitors act in a newly infected cell, the cell may be completely protected, as there is likely to be only one functional PIC in most infected cells. Integrase inhibitors might therefore protect cells for hours after plasma drug levels have dropped below optimal inhibitory levels.
While this laboratory evidence provides the rationale to perform a careful clinical trial of qd RAL, now underway with Merck sponsorship, it is far from proof that qd RAL is safe and effective. For example, especially during high-level viremia such as that seen prior to initial therapy, RT inhibitors may inhibit the production of DNA genomes and PICs in most infected cells, but not in all. If RAL is given once a day with 2 NRTIs, it will protect all cells in the first 12 hours, but the few cells that get infected in the second 12 hours, and are able create a PIC despite NRTIs, may not have enough RAL left inside to inactivate the PIC. Time and clinical trials will tell.
|
| |
|
|
|
|
|