 |
|
|
| |
Pharmacology at ICAAC-IDSA - The Maraviroc and Raltegravir Interaction: Do We Know How to Dose When Using These Two Important Agents?
|
| |
| |
Written by Stephen Becker, MD
Maraviroc (MVC) and raltegravir (RGV) are the two novel class agents recently approved by the FDA and EMEA. The initial use of these agents has been in treatment experienced patient populations where they provide a potent backbone for construction of a new regimen. Along with the PI darunavir and NNRTI etravirine, MVC and RGV offer the possibility of a suppressive regimen for most individuals with conventional three and four class resistance. Both agents have also successfully completed clinical trials in treatment naïve patients, and approval for this indication will be sought. While neither the metabolism of MVC or RGV, nor other known aspects of their pharmacology, would suggest an interaction between the drugs, a poster presented by the Pfizer group suggests such an interaction exists. It is the magnitude of this interaction, and the clinician's need to possibly adjust dosage of RGV that is the subject of this report.
Maraviroc is a substrate for the CYP450 3A4 isoenzyme. It does not meaningfully induce or inhibit 3A4 or other CYP450 enzymes. Raltegravir is metabolized not by the phase I CYP enzymes, but rather the conjugative enzyme UGT 1A1. Raltegravir does not inhibit or induce enzymes of the CYP450 system. Both MVC and RGV are substrates, but not inhibitors of the cellular efflux pump P-gp. Other aspects of the pharmacology (absorption, distribution and elimination) of MVC and RGV would not appear to explain any potential drug-drug interactions. Two features of RGV pharmacology however bear mention. First, to date there has been no robust pharmacokinetic-pharmacodynamic (PK-PD) relationship established. Data presented [1] from a pooling of phase II and phase III studies demonstrates shallow associations of Call and Cmin with virologic response, but other factors including the baseline HIV RNA and number of active agents in the regimen were more predictive of overall response. (Most agents, including MVC, have determined a PK parameter [often the Cmin or Cavg] associated with HIV RNA reduction.) The lack of a clear PK-PD relationship is not necessarily a concern, and may result from RGV exposures that are at the high end of the dose-response curve. Secondly, an unusually large inter- and intra-subject variability of plasma concentrations (in excess of 200% and 100%, respectively) is characteristic of RGV. As a point of comparison, the variability of most antiretrovirals is between 40% and 80%. The underlying reason explaining this large variability is not well understood.
The current interaction study [2] was performed by scientists from Pfizer. Enrolling 18 healthy volunteers (16 males, 2 females, 9 black, 6 white, 3 other self reported race) this open-label fixed sequence (non-cross-over) study evaluated RGV levels before and after the addition of MVC. Seventeen subjects completed the study and form the basis of the analysis. Both drugs were administered at conventional doses on an empty stomach. RGV was given for 3 days followed by a washout of 2 days. MVC was given alone for 6 days, and then the two agents were administered together for 3 additional days. PK samples were obtained at day 3 (RGV), day 11 (MVC) and day 14 (RGV and MVC). Validated assays were used to perform plasma drug levels and results were reported as geometric mean ratios (and 90% CI) for the Cmax, AUCt and C12. There was a slight reduction of MVC when coadministered with RGV as noted in table 1.
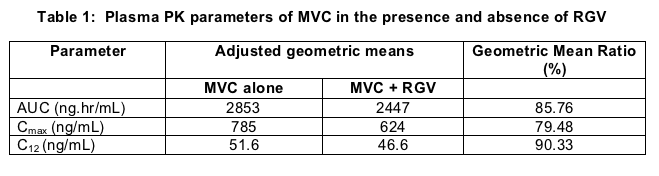
The noted reduction in exposure is not clinically significant, and no dose modification of MVC when dosed with RGV is needed.
Raltegravir concentrations were reduced when dosed with MVC (table 2). Mean C12 (concentration 12 hours following the last dose) was reduced by 28% (90% CI 9, 42), while AUC was reduced by 37%. Reductions of exposure greater than 25-30% are usually considered clinically significant, and in most cases prompt dosage alteration.
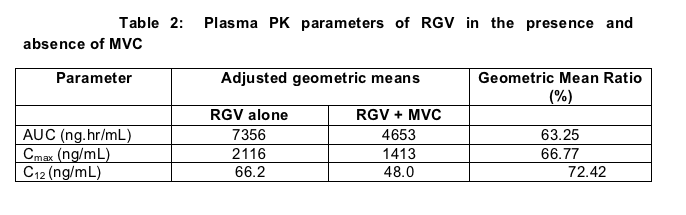
Two of the 17 subjects had striking reductions of RGV C12, one of 76% and the other of 69% (GMRs of 24% and 31%). Further when individual plots are examined it is evident that most subjects manifested a fall in C12 values when RGV was co-dosed with MVC; however 3 subjects had higher RGV values following co-administration. The reasons for the large and disparate inter-subject variability and the reason for variation in response to co-administration are not clear and require explication. Indeed the reason for - and mechanism of - the interaction is unknown. Similar to the unanticipated interactions of tenofovir with didanosine and several PIs, the eventual mechanism of the MVC-RGV interaction will likely expand our knowledge of pharmacology.
The clinical relevance of these findings lies in the potential importance of ensuring adequate plasma concentrations of RGV. As is widely appreciated, among treatment experienced individuals, RGV is a potent agent, and when combined with other active antiretrovirals successfully and durably suppresses viral replication. In the setting of multiple active agents variable RGV concentrations may not be significant. However should RGV be used as part of a regimen with reduced overall susceptibility, pharmacokinetic variability may prove clinically important. In this setting any further reduction of RGV concentrations consequent to co-administration with MVC may lead to virologic failure and drug resistance.
Several relevant questions are posed by this study. The mechanism of the apparent and unanticipated MVC-RGV interaction should be determined. In addition a more complete understanding of the factors responsible for the substantial variability of RGV exposures and current PK-PD relationship is important. At least two design features of the current study may contribute to the puzzling findings. First, given the considerable inter-subject variability of RGV, a larger sample size for future drug interaction studies is justified. Secondly a cross-over design would eliminate any effect of initial MVC dosing on the subsequent MVC-RGV interaction. In the mean time is RGV dose modification warranted? In settings where other agents of the regimen are anticipated to be fully active, no dosage alteration seems warranted. However where MVC and RGV are the primary active agents, an increase of RGV dose may be considered prudent. As is apparent to both patients and clinicians alike, the HIV pipeline beyond the recently approved agents is perilously thin. Every effort should be made to "protect" our important therapeutics, perhaps none more so than RGV. Further data is clearly needed, and Merck, the developer of RGV, should take the lead and provide the necessary clarity.
1. Wenning, Pharmacokinetic/Pharmacodynamic (PK/PD) analyses for raltegravir (RAL) in phase III studies in treatment experienced HIV- infected patients following 48 weeks of treatment; abstract H-4054
2. Andrews, A pharmacokinetic study to evaluate an interaction between maraviroc and raltegravir in health adults, abstract H-4055
|
| |
|
|
|
|
|