 |
|
|
| |
Pharmacokinetic-pharmacodynamic analyses of once-daily darunavir in the ARTEMIS study
|
| |
| |
Reported by Jules Levin
9th International Workshop on Clinical Pharmacology of HIV Therapy
April 7-9, 2008
New Orleans
Vanitha Sekar,1 Carline Vanden Abeele,2 Ben Van Baelen,2 Peter Vis,3 Ludo Lavreys,2 Martine De Pauw,2 Stephanie Dincq,2 Tony Vangeneugden,2 Sabrina Spinosa-Guzman,2 Richard Hoetelmans2
1Tibotec Inc., Yardley, PA, USA; 2Tibotec BVBA, Mechelen, Belgium; 3Exprimo NV, Mechelen, Belgium
Introduction
· Darunavir with low-dose ritonavir (DRV/r) at a dose of 600/100mg bid has been approved in the USA1 and in other countries2 for the treatment of HIV-1 infection in treatment-experienced adult patients.
· The ARTEMIS trial (TMC114-C211) was designed to compare the efficacy, safety and tolerability, resistance characteristics and pharmacokinetics of DRV/r at a dose of 800/100mg qd versus lopinavir/ritonavir (LPV/r) 800/200mg total daily dose in treatment-naive, HIV-1-infected patients.
· In ARTEMIS, 84% of DRV/r and 78% of LPV/r patients achieved the primary efficacy endpoint of viral load <50 copies/mL (intent-to-treat/time-to-loss of virologic response) at Week 48.3
· The current analysis explored the relationship between DRV pharmacokinetics and efficacy and safety following treatment with DRV/r 800/100mg qd in ARTEMIS.
Author Conclusions
In ARTEMIS, DRV/r 800/100mg qd was shown to be effective and had a favorable tolerability profile at Week 48.
The current analysis showed that DRV C0h was consistently above the protein binding-corrected EC50 value for wild-type virus of 55ng/mL in all patients. Furthermore, the median DRV C0h was 37-fold greater than this EC50 value.
No relevant relationships between DRV pharmacokinetics and efficacy or safety at Week 48 were observed in antiretroviral-naive, HIV-1-infected adult patients, at a given dose of DRV/r 800/100mg qd.
Program Abstract
Background: ARTEMIS is an ongoing Phase III study assessing the efficacy, safety and pharmacokinetics (PK) of darunavir with low-dose ritonavir (DRV/r) versus lopinavir with low-dose ritonavir (LPV/r) in treatment-naive HIV-1-infected adult patients. Patients were randomised to receive DRV/r 800/100mg once daily (n=343) or LPV/r 800/200mg total daily dose (n=346). In addition, all patients received a fixed daily-dose of emtricitabine and tenofovir. In the primary analysis of this study, 84% of DRV/r and 78% of LPV/r patients achieved HIV-1 RNA <50 copies/mL (TLOVR) at Week 48. The current analysis explored the relationship between DRV PK and efficacy and safety following treatment with DRV/r 800/100mg once daily in the ARTEMIS study.
Material & Methods: Sparse blood sampling was performed for 335 patients randomised to the DRV/r arm to determine steady-state area under the curve (AUC24h) and predicted trough concentration (C0h) for DRV using a population pharmacokinetic model. Relationships between DRV PK and efficacy at Week 48 were assessed using analysis of covariance models. Efficacy measurements tested in the model included change in HIV-1 RNA from baseline and proportion of patients achieving HIV-1 RNA <50 copies/mL. The relationships between DRV PK and safety, and the occurrence of adverse events of interest, were also investigated using descriptive methods.
Results: The plasma trough concentration (C0h) of DRV exceeded the in-vitro protein binding-corrected EC50 of 55 ng/mL for wild-type virus in all 335 patients evaluated (range: 368-7,242 ng/mL). No relationships were observed between DRV PK and efficacy response parameters. Changes in HIV-1 RNA from baseline at Week 48 by DRV AUC24h quartile ranges of ≦ 75,598; 75,599-87,854; 87,855-103,840 and >103,840 ng.h/mL were -3.11, -3.17, -3.07 and -3.11 log10 copies/mL, respectively. For these AUC24h quartile ranges of DRV, the proportions of patients reaching HIV-1 RNA <50 copies/mL were 94%, 95%, 90% and 93% (observed data), respectively. No apparent relationships were observed between DRV PK parameters and the occurrence of rash, nervous system and psychiatric disorders, or cardiac, gastrointestinal, liver, lipid and glucose-related adverse events.
Conclusion: No relevant relationships between DRV PK and efficacy or safety at Week 48 were observed at a DRV/r dose of 800/100mg once daily in treatment-naive HIV-1-infected adult patients.
Results
Patient characteristics
· A total of 689 patients (n=343 in the DRV/r arm and n=346 in the LPV/r arm) were randomized and treated.
· Baseline characteristics were well-balanced between treatment arms. The trial included a diverse population where women and non-Caucasians were well represented (30% and 58%, respectively).
· Consistent with the treatment-naive status of patients, few patients (9%) had CDC category C HIV infection. At baseline, 34% of patients had >100,000 copies/mL and median CD4 cell count was 225 cells/mm3.
Pharmacokinetics
· Of the 343 patients randomized and treated with DRV/r, 335 patients with sparse sampling data were included in the population PK analysis for DRV.
· Figure 1 and Table 1 show the Bayesian estimates of the DRV PK parameters from the sparse sampling. For all DRV/r-treated patients, DRV C0h was consistently above 55ng/mL, which is the protein binding-corrected EC50 value for wild-type virus. The median DRV C0h was 37-fold greater than this EC50 value.
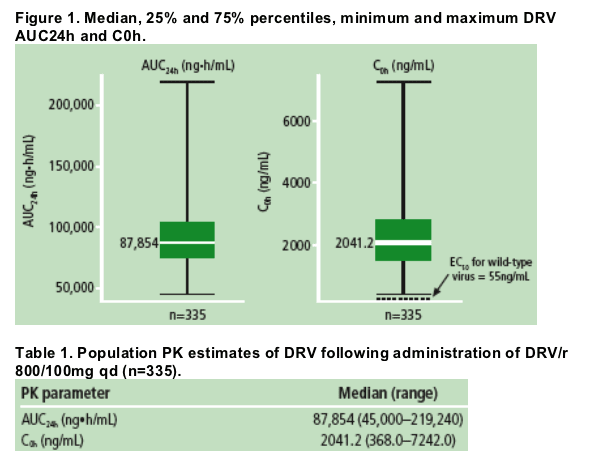
Relationship between pharmacokinetics and efficacy
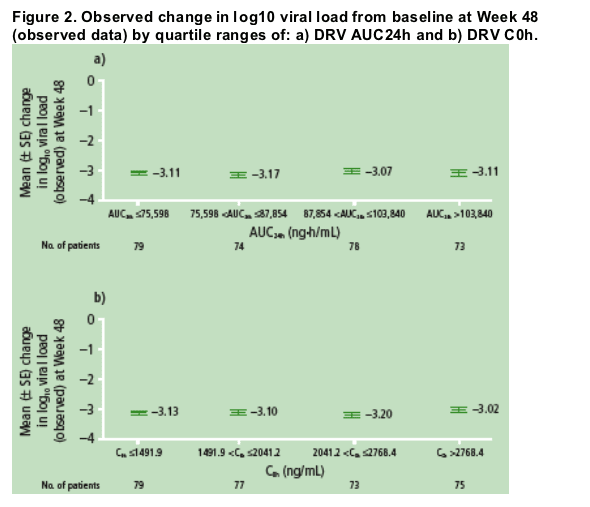
No apparent relationships were observed between DRV AUC24h
and C0h quartile ranges and observed change in log10 viral load
from baseline at Week 48 (Figure 2).
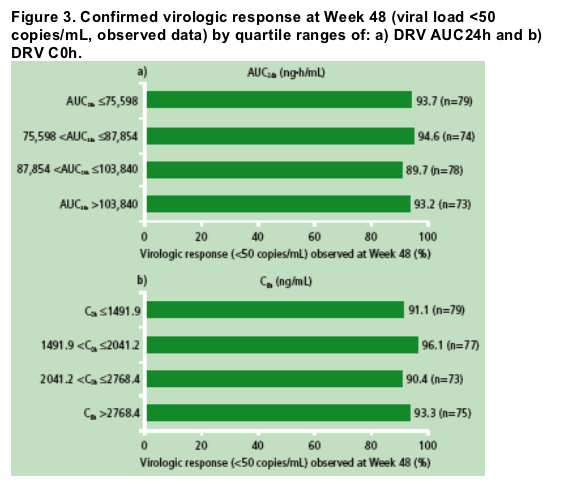
There was no relevant relationship between DRV AUC24h and C0h and observed proportion of patients achieving plasma viral load <50 copies/mL at Week 48 (Figure 3).
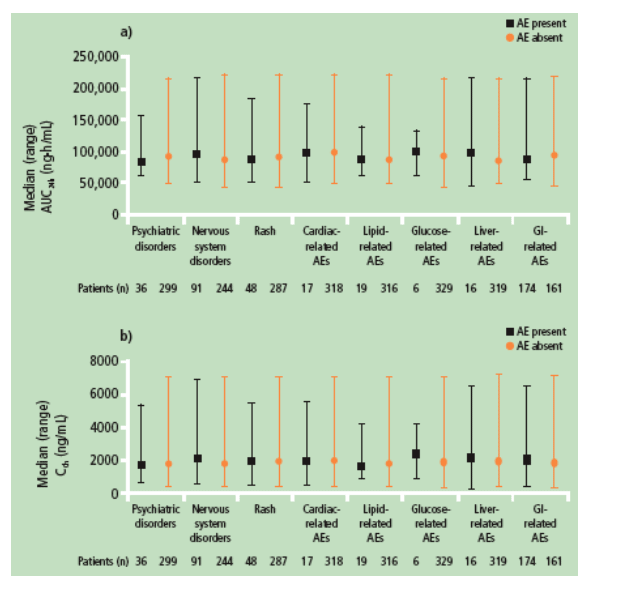
Relationship between pharmacokinetics and safety
No apparent relationships were observed between DRV AUC24h and C0h and occurrence of rash, nervous system and psychiatric disorders, or cardiac, GI, liver, lipid and glucose-related AEs (Figure 4).
Methods
Patients and study design
· In the ongoing, randomized, controlled, open-label, Phase III ARTEMIS trial, treatment-naive, HIV-1-infected, adult patients were randomized to receive DRV/r 800/100mg qd or LPV/r 800/200mg total daily dose, plus a standard daily-dose of tenofovir disoproxil fumarate/emtricitabine.
· The study protocol was reviewed and approved by the appropriate institutional ethics committee(s) and health authorities, and the study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients.
Pharmacokinetic (PK) and pharmacodynamic (PD) analysis
· Sparse sampling for the determination of plasma concentrations of DRV and ritonavir was performed in all patients
- blood samples were drawn at Weeks 4, 8, 24, 48, 72 and 96, with two samples taken at Weeks 4 and 24 (the first sample immediately before DRV/r intake, and the second at least 1 hour after the first was drawn).
· Plasma concentrations of DRV and ritonavir were determined by a validated liquid chromatography-mass spectrometry/mass spectrometry method. The lower limit of quantification was 10.0ng/mL for DRV and 5.00ng/mL for ritonavir.
· Estimates of exposure (area under the curve; AUC) and trough concentrations (C0h) of DRV were calculated to perform PK/PD analyses
- a population PK model was developed for DRV on the basis of data in HIV-1-infected patients and healthy volunteers.4 This model was then applied to the samples drawn in this trial to derive empirical Bayesian estimates of DRV exposure at steady-state
- the final population PK model to describe the pharmacokinetics of DRV in this trial was a two-compartmental model with first-order absorption. Apparent total clearance was modeled to be dependent on concentrations of alpha-1 acid glycoprotein (AAG), with a decreasing clearance with higher concentrations of AAG
- individual estimates of AUC24h and C0h were obtained at each visit where plasma samples were obtained, and a median value for each parameter was calculated for each individual patient from these values.
· Relationships between DRV AUC24h and C0h and virologic efficacy at Week 48 were assessed using analysis of covariance models. Efficacy measurements tested in the model included change in log10 viral load from baseline and proportion of patients achieving viral load <50 copies/mL (observed data used for both measurements).
· For relationships between DRV PK and safety, descriptive statistics of DRV AUC24h and C0h were used for patients who did or did not present with the following adverse events (AEs): rash-related, cardiac-related, gastrointestinal (GI)-related, liverrelated, lipid-related, glucose-related, psychiatric and nervous system-related AEs.
References
1. Tibotec Inc. PREZISTATM (darunavir) Prescribing Information. October 2006 [accessed January 3, 2008]. Available from: http://www.prezista.com.
2. PREZISTATM (darunavir) Summary of Product Characteristics. February 2007 [accessed January 3, 2008]. Available from: http://www.emea.europa.eu/humandocs/PDFs/EPAR/prezista/H-707-PI-en.pdf.
3. DeJesus E, et al. 47th Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, September 17-20 2007. Abstract H-718b.
4. Vis P, et al. 15th Meeting of the Population Approach Group in Europe, Bruges, Belgium, June 14-16 2006. Abstract 964.
|
| |
|
|
|
|
|