 |
|
|
| |
Integrase Inhibitor Resistance (elvitegravir/raltegravir) Involves Complex Interactions Among Primary and Secondary Resistance Mutations:
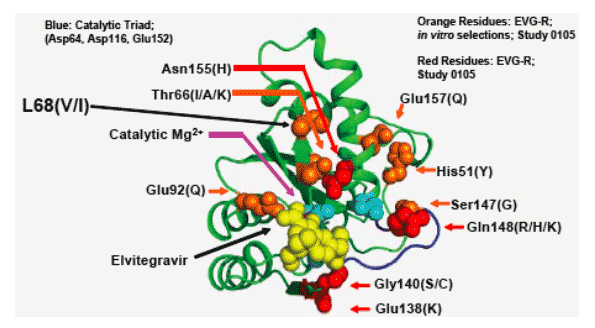
A Novel Mutation L68V/I Associates with E92Q and Increases Resistance
|
| |
| |
Reported by Jules Levin
XVII HIV Drug Resistane Workshop
June 10-14, 2008
Sitges, Spain
DD Goodman,1 R Hluhanich,2 J Waters,1 NA Margot,2 S Fransen,3 S Gupta,3 W Huang,3 N Parkin,3 K Borroto-Esoda,1 ES Svarovskaia,1 MD Miller,2 and DJ McColl2
1Gilead Sciences, Inc., Durham, NC, USA; 2Gilead Sciences, Inc., Foster City, CA, USA; 3Monogram Biosciences, Inc., South San Francisco, CA, USA
AUTHOR CONCLUSIONS
Resistance to INIs involves complex interactions among both primary and secondary INI-R mutations
- Evidence of mutual exclusions between primary INI-R mutations,
(E92Q and Q148R, and Q148R and N155H), were observed in this study
INI-R mutations generally reduce viral fitness compared to wild-type
- The single primary INI-R mutants analyzed had relative fitness of
WT > E92Q > N155H > Q148R/H/K
E92Q and N155H appear to be compatible with one another and amplify resistance to EVG and RAL, but relative viral fitness decreases for the double mutant
- Double mutants of Q148R with either E92Q or N155H may not be observed clinically due to more severely reduced viral fitness
L68V associates specifically with E92Q in patient isolates and enhances resistance to both EVG and RAL when combined with E92Q
- L68V does not compensate for the reduced relative fi tness of E92Q, Q148R or N155H IN mutations
- Lack of clinical association of L68V with N155H or Q148R may be due to the lack of significantly increased INI resistance when combined with N155H or lack of a compensatory effect for the more fitness impaired Q148R mutation
Secondary INI-R mutations can either enhance resistance (e.g. L68V) or may both enhance resistance and partially compensate for reduced viral replication (e.g. G140S, S147G)
- S147G can partially compensate for reduced replication of Q148R, but not that of E92Q6
Acquisition of increased INI phenotypic resistance with maintenance of adequate or improved viral fitness is a major driver of viral evolution among the diverse pathways of genotypic resistance to INIs
INTRODUCTION
Human immunodeficiency virus (HIV) encodes three enzymes essential for viral
replication: reverse transcriptase (RT), protease (PR) and integrase (IN)
An HIV-1 integrase strand transfer inhibitor, raltegravir (RAL), has recently been approved for use in antiretroviral (ARV) therapy-experienced patients
Elvitegravir (EVG, GS-9137/JTK-303) is another potent HIV integrase strand transfer inhibitor (serum-free antiviral IC50 = 0.2 nM; IC90 = 1.2 nM in PBMCs)1 EVG is active against HIV-1 isolates resistant to all NRTIs, NNRTIs and PIs1
A phase 2 dose-ranging study of EVG (Study 0105) has investigated EVG as a once-daily, ritonavir-boosted therapy (EVG/r) in ARV-experienced HIV-1 infected patients2
IN genotypic and phenotypic resistance patterns from patients experiencing virologic failure (VF) on EVG/r 125 mg in Study 0105 have been described3
In this study, linkage of IN mutations, phenotypes of clinically relevant IN mutants and viral replication and relative fitness of IN mutants were studied
OBJECTIVES
Characterize linkage among IN mutations in clinical viral isolates from Study 0105 using single genome sequencing (SGS) of plasma HIV-1 and clonal analysis of resistance test vectors in the PhenoSense INI assay
Characterize the phenotypic susceptibility to EVG, RAL and control ARV compounds of HIV-1 site-directed IN mutants
Characterize the impact of IN mutations on relative fitness (multi-cycle growth competition, 1+s) and viral replication capacity (single-cycle growth in PhenoSense INI Assay)
METHODS
Study Design: Study 0105 was a phase 2, randomized, active control, partially blinded (dose of EVG) dose-ranging 48 week study. Study 0105 was a non-inferiority study of RTV-boosted EVG (EVG/r) versus RTV-boosted comparator PI (CPI/r) in combination with an optimized background therapy (OBT) in ARV-experienced, HIV-1 infected patients. OBT initially included only NRTIs +/-enfuvirtide (T-20) (used at the investigator's discretion). Patients were randomized 1:1:1:1 to either 20, 50 or 125 mg EVG/r (boosted with 100 mg RTV) or to the CPI/r arm. The PIs DRV or TPV were permitted after Week 8.
Virologic Failure (VF): VF was defined as HIV-1 RNA > 400 copies/mL and < 1 log10 reduced from baseline by Week 12 subsequently confi rmed; OR, at any time after Week 16, confi rmed rebound to < 1 log10 from baseline and > 400 copies/mL HIV-1 RNA. IN genotype/phenotype data were obtained on 28/30 VF subjects from the EVG/r 125 mg arm.
Single Genome Sequencing (SGS): SGS of plasma HIV-1 from VF subjects used the methods previously described4. Dideoxy sequencing covered the entire IN gene (amino acids 1-288). The amino acid sequence generated by each clone was compared to an HIV-1 Clade-B reference.
Construction of HIV-1 Site-Directed IN Mutants and Phenotyping: Site-directed mutagenesis and production of mutant virions in HXB2 was as previously described3. Infection of MT-2 cells with viruses and phenotypic testing was as previously described3. Cell viability in antiviral assays was detected using chemiluminescence (Cell Titer-Glo); data were converted to "% cell death"; antiviral IC50s were determined using curve fitting (GraphPad, Prism). Phenotypes were expressed as fold change relative to the HXB2 reference.
Competitive Relative Fitness Assay (1+s): Site-directed mutant viruses were
subcloned into two LAI-based vectors containing silent marker mutations. Multi-cycle growth competition experiments were performed in MT-2 cells. MOI used was <0.1 throughout the competition experiment to minimize recombination. Viral replication was monitored by allele-specifi c real-time RT-PCR at Day 3, 6, and 9 after infection. Relative fitness value was calculated as previously described: (1+s) = exp [1/t &%176;- Ln (Mt2/Wt2 &%176;- Mt1/Wt1)], where t is time in days; Mt2, Mt1, Wt2, and Wt0 are fractions of mutant virus at two time points of a measurement and fractions of wild-type virus at two time points of a measurement, respectively5.
GeneSeq INI and PhenoSense INI Assay (Monogram Biosciences): Individual IN resistance test vector clones from VFs on EVG/r 125 mg were sequenced to define linkage of IN mutations and their effect on EVG susceptibility as previously described3.
IN Replication Capacity: The effect of IN mutations on viral replication capacity was assessed using individual resistance test vector clones in the PhenoSense INI assay as previously described3.
RESULTS
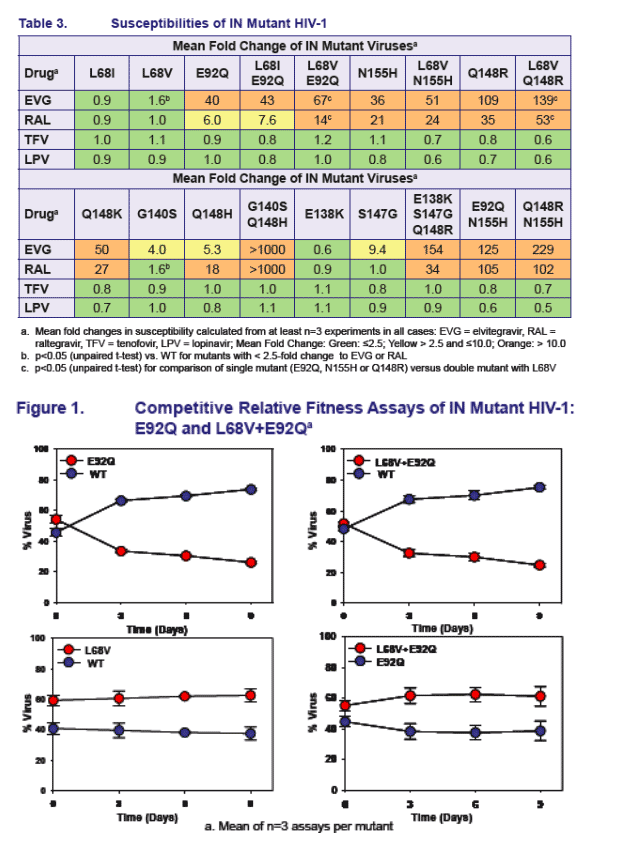
Population genotypes showed an association of L68V/I with E92Q (6/6 with L68V/I)
SGS analysis showed that L68V/I was on the same viral genomes as E92Q (Table 2)
- L68V+E92Q signifi cantly amplifi ed resistance to both EVG and RAL, compared to E92Q alone (Table 3)
Although not observed together in clinical isolates, site-directed mutants showed that L68V significantly amplifi ed resistance to EVG and RAL when added to Q148R, but only marginally increased resistance when added to N155H (Table 3)
SGS analysis of patients with either E92Q+N155H or E92Q+Q148R population
genotypes showed that E92Q and N155H mutations could be found on the same viral genome but not E92Q and Q148R mutations (Table 2)
- Infectivity (TCID50) of E92Q+Q148R and Q148R+N155H viruses was low (not shown)


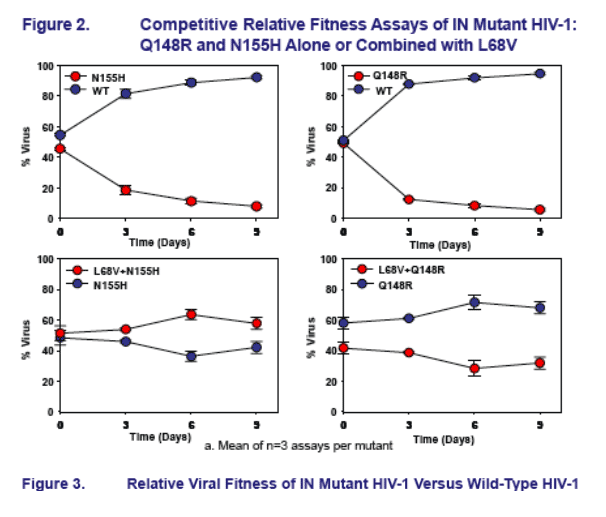
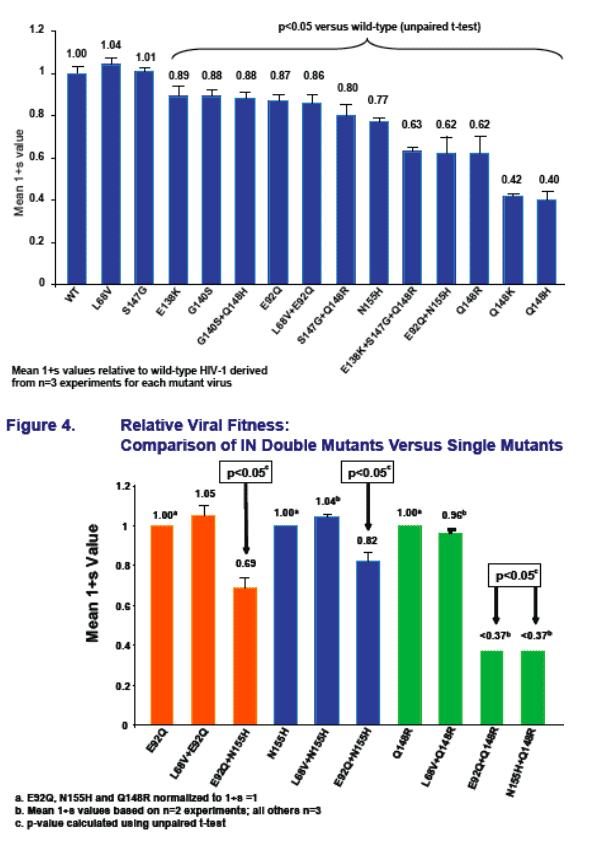
All INI-R viruses, except L68V and S147G, had significantly reduced relative fitness (1+s) versus wild-type HIV-1
Among the primary INI-R mutations, relative fitness versus wild-type HIV-1 followed the order WT>E92Q>N155H>Q148R/K/H
L68V did not compensate for reduced relative fi tness of either E92Q, Q148R or N155H
- L68V appears to be a secondary INI-R mutation, signifi cantly increasing resistance when combined with E92Q but not N155H
- L68V did not compensate for the signifi cantly reduced fi tness of Q148R but did amplify resistance
- Mutations G140S and S147G can both partially compensate for reduced fitness of Q148R/H/K and amplify INI resistance when added to Q148R/H/K
The E92Q+N155H mutant had significantly reduced relative fi tness but increased resistance to both EVG and RAL, compared to either single mutant
Mutants E92Q+Q148R and Q148R+N155H show signifi cantly reduced relative fitness when compared to Q148R
- Infectivity of E92Q+Q148R mutant was too low for INI phenotyping to be achieved (not shown)
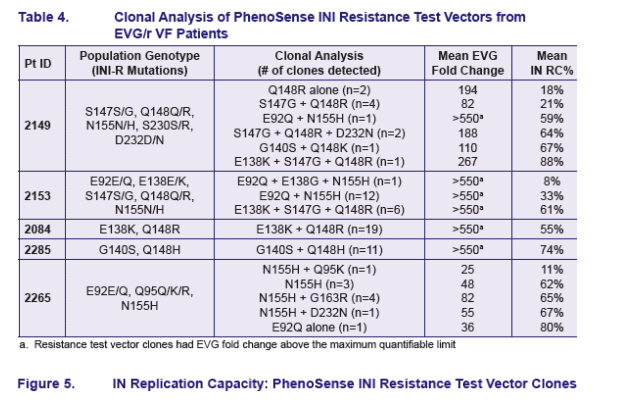
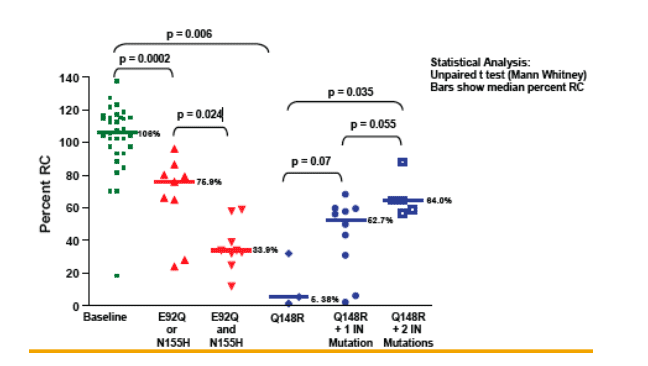
Clonal analysis of EVG/r patients with complex IN genotypes showed evidence of the presence of multiple viral quasi-species
RC of IN mutants followed the order E92Q>N155H>Q148R
E92Q+N155H mutations were found on single clones but not E92Q+Q148R or
Q148R+N155H mutations; the latter were detected together only in population genotypes
E92Q+N155H had decreased RC but increased EVG resistance relative to either
mutation alone
Resistance test vectors with Q148R alone had lower RC than those with Q148R plus other IN mutations suggesting compensatory effects in the latter groups
Mutations E138K, G140S and the mutants E138K+S147G increased IN RC and tended to increase resistance to EVG when added to viruses carrying the Q148R mutation
REFERENCES
1. Matsuzaki, et al 2006; Poster #508, 13th Conference on Retroviruses and Opportunistic Infections, Denver, CO
2. Zolopa, et al 2007, Oral Presentation 143LB, 14th Conference on Retroviruses and Opportunistic Infections, Los Angeles, CA
3. McColl, et al 2007, Oral Presentation #9, 16th International HIV Drug Resistance Workshop, Barbados
4. Palmer, et al 2005, J. Clin. Micro.; 45: 406-413
5. Wu, et al 2006 J Virol.; 80:2380-2389
6. Cavanaugh, et al 2007; Poster #32, 8th Annual Symposium on Antiviral Drug Resistance Targets and Mechanisms, Richmond, VA
|
| |
|
|
|
|
|