 |
|
|
| |
Quantification of HIV Tropism by "Deep" Sequencing Shows a Broad Distribution of Prevalence of X4 Variants in Clinical Samples that Is Associated with Virological Outcome
|
| |
| |
Reported by Jules Levin
CROI 2009 (SEE POSTER DETAILS FOLLOWING ABSTRACT BELOW)
"A low prevalence of X4 was associated with improved virological response to maraviroc, even where standard Trofile indicated DM or X4 virus."
L Swenson1, W Dong1, T Mo1, C Woods1, A Thielen2, M Jensen3, C Glascock1, J Montaner1,4, and Richard Harrigan*1,4
1BC Ctr for Excellence in HIV/AIDS, Vancouver, Canada; 2Max-Planck-Inst for Informatics, Saarbrucken, Germany; 3Fortinbras Res, Buford, GA, US; and 4Univ of British Columbia, Vancouver, Canada
Background: "Deep" sequencing (Roche GS-FLX) detects minority HIV variants in clinical samples. Here, we performed both deep and standard sequence analyses of the HIV V3 region from individuals entering a clinical trial of maraviroc, all of whom had evidence of X4/DM virus and would not be expected to show a virological response.
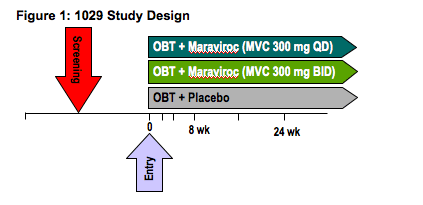
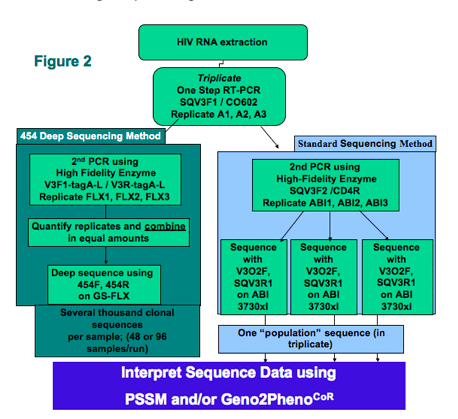
Methods: Screening samples from the Pfizer A4001029 study were polymerase chain reaction (PCR) amplified in triplicate (n = 202). Tropism phenotype of all samples was X4 or DM by the standard Monogram Trofile assay as defined by the study entry criteria. Conventional (n = 153) and "deep" sequencing using the GS-FLX was performed using a "barcoding" approach, allowing the simultaneous analysis of 48 samples in both directions (n = 202) in a blinded manner. V3 genotypes were interpreted using the PSSM and/or geno2pheno algorithms.
Results:
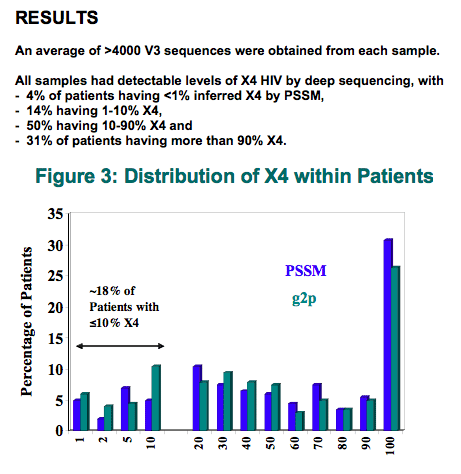
An average of >4000 V3 sequences were obtained from each sample.
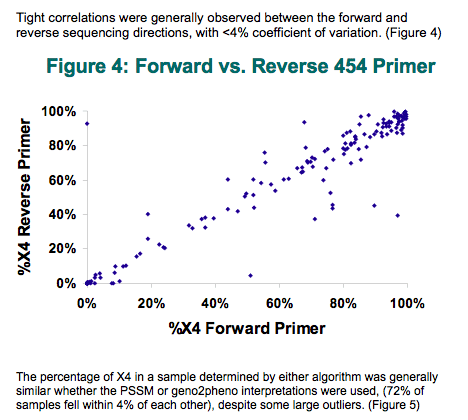
All samples had detectable levels of X4 HIV by deep sequencing, with 4% of patients having ≤1% inferred X4 by PSSM, 14% having 1 to 10% X4, 50% having 10 to 90% X4, and 31% of patients having more than 90% X4. Tight correlations were generally observed between the forward and reverse sequencing directions, with <4% coefficient of variation.
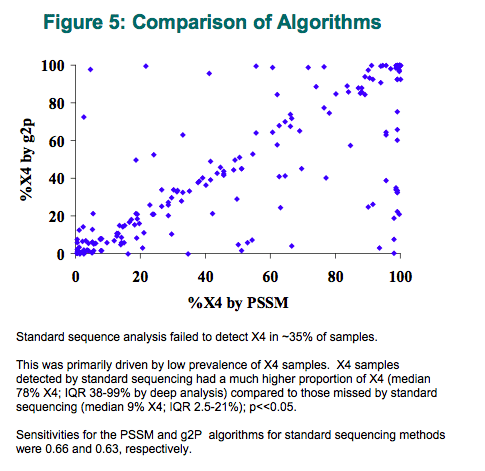
The percentage of X4 determined was generally similar whether the PSSM or geno2pheno interpretations were used, (72% of samples fell within 4% of each other), despite some large outliers. Standard sequence analysis failed to detect X4 in 28% of samples. This was primarily driven by the low prevalence of X4-samples which standard sequence detected as having X4 virus had a much higher proportion of X4 (median 78% X4; IQR 38 to 99% by "deep" analysis) compared to those missed by standard sequencing (median 9% X4; IQR 2.5 to 21%); p <<0.05.
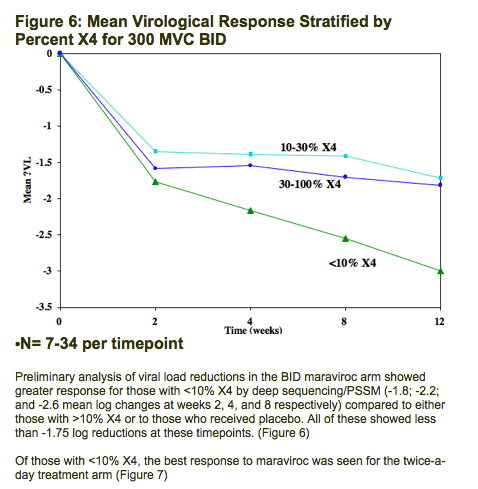
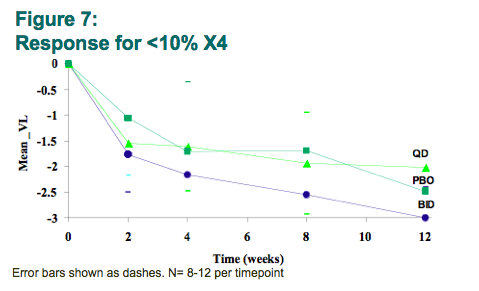
Preliminary analysis of viral load reductions in the twice-daily maraviroc arm showed greater response for those with <10% X4 by deep sequencing/PSSM (-1.8, -2.2, and -2.6 mean log changes at weeks 2, 4, and 8, respectively) compared to either those with >10% X4 or to those who received placebo. All of these showed <-1.75 log reductions at these points.
Conclusions:
Deep sequence analyses detect and quantify low prevalence of X4 HIV within clinical isolates that are not detected by standard sequence analysis. A low prevalence of X4 was associated with improved virological response to maraviroc, even where standard Trofile indicated DM or X4 virus.
Quantification of HIV Tropism by "Deep" Sequencing Shows a Broad Distribution of Prevalence of X4 Variants in
Clinical Samples Associated with Virological Outcome
P. Richard Harrigan1,4 Luke C. Swenson1, Winnie Dong1, Theresa Mo1, Conan Woods1, Alexander Thielen2, Mark Jensen3, Christopher Glascock1, & Julio S Montaner1,4
1BC Centre for Excellence in HIV/AIDS, Vancouver, Canada; 2Max-Planck Institute for Informatics, Saarbrucken, Germany; 3Fortinbras Research, Buford, GA, 4 Faculty of Medicine, University of BC, Vancouver Canada
BACKGROUND
Deep sequencing (Roche GS-FLX) detects minority HIV variants in clinical samples. Here, we performed both standard and "deep" sequence analyses of the HIV V3 region from individuals entering a clinical trial (Pfizer A4001029) of the CCR5-inhibitor, maraviroc. All patients entering the trial had previous antiretroviral experience, viral loads >1000 HIV RNA copies/mL and evidence of X4 or DM virus by the standard Monogram assay. Overall, patients would not be expected to show a significant virological response to maraviroc.
AUTHOR CONCLUSIONS
Deep sequence analyses detect and quantify low prevalences of X4 HIV within clinical isolates.
Samples with low prevalence were commonly missed by standard V3 sequencing.
Preliminary analysis suggests that a minority of patients with a low proportion of X4 virus may be associated with early virological response to maraviroc, despite being called DM by standard Trofile.
METHODS
Screening samples from the 1029 study were PCR amplified in triplicate (N=202). Tropism phenotype of all samples was X4 or DM by the standard Monogram Trofile assay as defined by the study entry criteria.
Conventional sequencing (N=153) was done using the ABI 3730. Deep sequencing using the GS-FLX was performed using a barcoding approach, allowing the simultaneous analysis of 48 samples in both directions (N=202) in a blinded manner. V3 genotypes were interpreted using the PSSM and/or geno2pheno algorithms.

|
| |
|
|
|
|
|