 |
|
|
| |
Antiviral activity and safety of TMC435 combined with peginterferon a-2a and ribavirin in patients with genotype-1 hepatitis C infection who failed previous IFN-based therapy
|
| |
| |
EASL April 23-26 2009 Copenhagen Denmark
Reported by Jules Levin
P. Marcellin,1 H. Reesink,2 T. Berg,3 M. Cramp,4 R. Flisiak,5 H. Van Vlierberghe,6 R.Verloes,7 O. Lenz,7 M. Peeters,7 V. Sekar8 and G. De Smedt7
1Hôpital Beaujon, Clichy, France; 2Amsterdam Medical Center, The Netherlands; 3Charite Campus Virchow Klinikum, Berlin, Germany; 4Derriford Hospital and Peninsula Medical School, Plymouth, UK;
5Medical University of Bialystok, Poland; 6University Hospital Ghent, Belgium; 7Tibotec BVBA, Mechelen, Belgium; 8Tibotec Inc., Yardley, PA, USA

INTRODUCTION
A substantial proportion of patients infected with hepatitis C virus
(HCV) genotype 1 do not achieve a sustained virologic response (SVR)
after combination therapy with pegylated interferon (PEGIFN) and
ribavirin (RBV).1
Better treatment options are required for treatment-experienced
patients: in a recent international, multicentre study, SVR response
rates to PEGIFN and RBV were only 10% in genotype-1 prior
non-responders and 27% in genotype-1 prior relapsers.2
TMC435 is a macrocyclic and selective HCV NS3/4A protease inhibitor
with an in vitro EC50 value of 8 nM in a genotype-1b replicon cell line.3
In Phase I studies, TMC435 was generally safe and well tolerated, and
the pharmacokinetic profile supported a once-daily (QD) dosing regimen.
A median decrease of 3.9 log10 IU/mL in HCV RNA was observed after five
days of monotherapy with TMC435 200 mg QD in genotype-1-infected
individuals who failed previous IFN-based therapies.3-5
OPERA-1 (TMC435350-TiDP16-C201) is a double-blind, placebocontrolled,
Phase IIa, proof-of-concept trial to assess the antiviral activity,
safety and pharmacokinetics of once-daily regimens of TMC435 in
HCV genotype-1 treatment-naïve and treatment-experienced patients.
Here we report the four-week results of OPERA-1 Cohort 4, for
treatment-experienced patients receiving TMC435 in combination
with PEGIFN and RBV.
METHODS
Study design
· The study design for OPERA-1 is summarised in Figure 1.
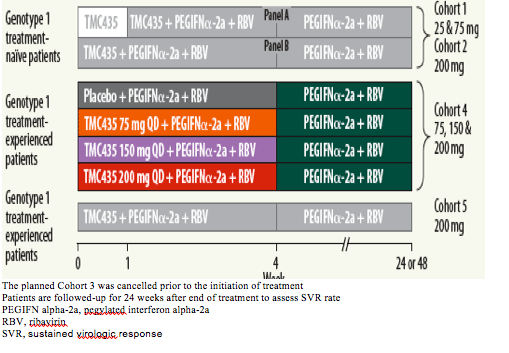
The planned Cohort 3 was cancelled prior to the initiation of treatment
Patients are followed-up for 24 weeks after end of treatment to assess SVR rate
PEGIFN alpha-2a, pegylated interferon alpha-2a
RBV, ribavirin
SVR, sustained virologic response
At the start of the double-blind treatment period of Cohort 4, patients were randomised to receive either TMC435 75 mg QD, 150 mg QD, 200 mg QD or placebo as part of a triple therapy regimen, in combination with pegylated interferon a-2a (PEGIFNa-2a) (180 µg subcutaneously, once weekly) and RBV (1000-1200 mg BID, body-weight dependent) for 28 days, followed by PEGIFNa-2a and RBV alone for a total treatment duration of 48 weeks.
Patient population
The treatment-experienced patients enrolled in this part of the study were:
- prior non responders: <2 log10 IU/mL decrease from baseline in HCV RNA after 12 weeks of prior IFN-based therapy, or
- prior relapsers: confirmed detectable HCV RNA after achieving undetectable HCV RNA at the end of treatment.
Eligible patients were: aged 18-70 years with documented chronic HCV (genotype 1; diagnosis >6 months prior to screening); required to have HCV plasma RNA of >10,000 IU/mL at screening; required to have compensated liver disease.
Key exclusion criteria included: receipt of a polymerase inhibitor, protease inhibitor or dual therapy with PEGIFN and RBV during the six months prior to screening; co-infection with HIV-1 or HIV-2, hepatitis A or B, or active tuberculosis at screening.
Study assessments
Antiviral activity
The primary efficacy objective was to determine the change from baseline in HCV RNA levels at Day 28. Serum samples were obtained at baseline and on Days 1 (4 h and 10 h), 2, 3, 7, 8, 11, 14, 21 and 28, and HCV RNA levels were quantified using the Roche COBAS TaqManHCV/HPS assay v2.0.
Other efficacy endpoints were to evaluate the response to treatment (HCV RNA below the lower limit of detection, <10 IU/mL and the lower limit of quantification, <25 IU/mL) and to assess the number of viral breakthroughs (>1 log10 IU/mL increase from nadir or HCV RNA levels >100 IU/mL in patients with previous HCV RNA levels <10 IU/mL).
Safety and tolerability
Safety and tolerability were monitored continuously throughout the trial. Vital signs, electrocardiogram (ECG) recordings and clinical laboratory tests were taken on Days 1, 7, 14, 21 and 28.
Statistical analysis
A pre-planned intent-to-treat analysis was performed when all patients had either completed 28 days of treatment or had discontinued earlier. All data are presented descriptively.
RESULTS
Baseline demographics and characteristics
In total in Cohort 4, 37 treatment-experienced patients were randomised to the four treatment groups: 25 prior non-responders and 12 prior relapsers to previous IFN-based therapy for HCV. All patients completed treatment to Day 28.
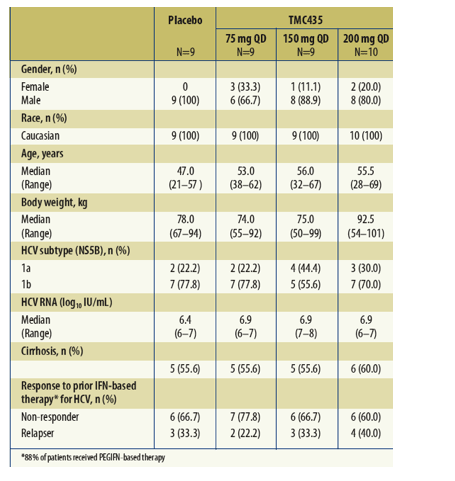
Patient demographics and baseline characteristics are shown in Table 1.
Table 1. Patient demographics and baseline characteristics.
Antiviral activity
Change in plasma HCV RNA from baseline
Mean decreases in plasma HCV RNA from baseline were 4.3, 5.5 and 5.3 log10 IU/mL for the TMC435 75, 150 and 200 mg QD groups, respectively, at Day 28, compared with 1.5 log10 IU/mL in the placebo group.
Mean decreases in plasma HCV RNA over time for each treatment group are shown in Figure 2.
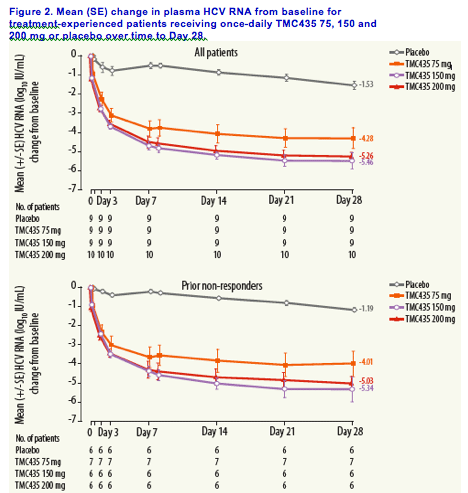
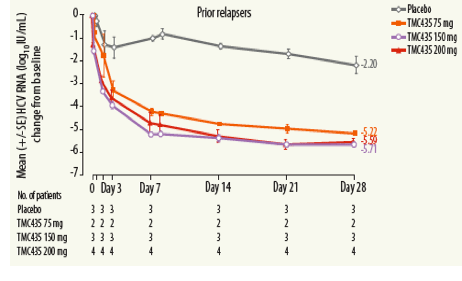
Week-4 virologic response
At Day 28, 4/9 (44%), 7/9 (78%) and 7/10 (70%) patients achieved plasma
HCV RNA levels <25 IU/mL in the 75, 150 and 200 mg QD treatment
groups, respectively, compared with no patients (0/9) in the placebo group.
The proportion of patients achieving plasma HCV RNA <10 IU/mL and
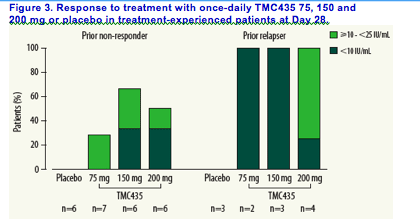
<25 IU/mL for both prior non-responders and relapsers is shown in
Figure 3.
Viral breakthrough
Three viral breakthroughs were observed, exclusively in patients infected with HCV genotype 1b who were prior non-responders to treatment; two in the 75 mg QD group and one in the 150 mg QD group.
At the time of the confirmed viral breakthrough, a D168V mutation in the NS3 protease domain was detected in all three patients using standard population sequencing.
Safety and tolerability
A summary of adverse events (AEs) is shown in Table 2.
Table 2. Summary of adverse events (AEs).
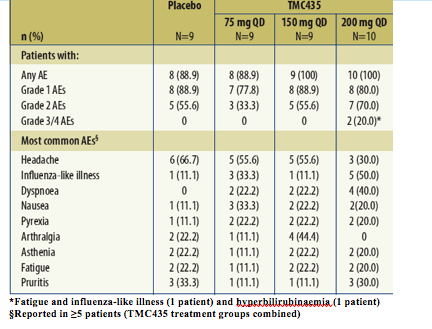
There were no discontinuations due to AEs, serious AEs or deaths during the study.
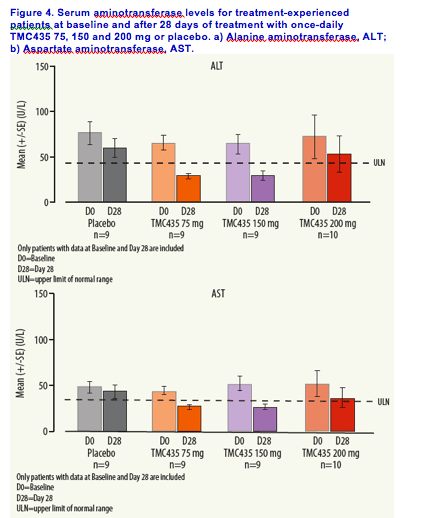
Alanine aminotransferase and aspartate aminotransferase levels decreased over time in all TMC435 treatment groups (Figure 4.).
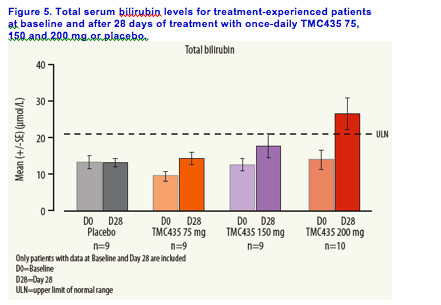
Bilirubin elevations were observed in some patients receiving TMC435
(total, direct and indirect), mostly with the 200 mg dose. These elevations were generally mild and reversible in nature.
No dose trends or clinically relevant changes were noted in any other
laboratory parameters, ECG parameters or vital signs.
References
1. Ghany MG et al. Diagnosis, management, and treatment of hepatitis C: An update. Hepatology 2008 2; 49:1335-1374.
2. Poynard T et al. Peginterferon alfa-2b and ribavirin: effective in patients with hepatitis C who failed interferon alfa/ribavirin therapy. The Epic Study Group. Gastroenterology 2009 Jan 22. [Epub ahead of print]
3. Lin TI et al. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob Agents Chemother 2009; 53:1377-1385.
4. Reesink H et al. Presented at the 43rd EASL, Milan, Italy, April 23-27, 2008, Oral presentation.
5. Van't Klooster G et al. Presented at the 59th AASLD, San Francisco, CA, October 31-November 4, 2008, Poster 1895.
|
| |
|
|
|
|
|