| |
Efficacy and safety of entecavir in nucleoside-naive, chronic hepatitis B patients:
Phase II clinical study in Japan
|
| |
| |
Journal of Gastroenterology and Hepatology
Volume 24, Issue 2, Pages 255-261
Published Online: 22 Jan 2009
Haruhiko Kobashi,* Kouichi Takaguchi, Hiroshi Ikeda, Osamu Yokosuka, Mitsuhiko Moriyama, Fumio Imazeki, Masayoshi Kage,** Taku Seriu, Masao Omata, Kousaku Sakaguchi* and Yasushi Shiratori*
*Department of Gastroenterology and Hepatology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Department of Internal Medicine, Kagawa Prefectural Central Hospital, Kagawa, Department of Gastroenterology and Hepatology, Kurashiki Central Hospital, Okayama, Department of Medicine and Clinical Oncology, Graduate School of Medicine, Chiba University, Chiba, Department of Gastroenterology and Hepatology, Nihon University Itabashi Hospital, Tokyo, **Department of Pathology, School of Medicine, Kurume University, Fukuoka, Bristol-Myers Squibb Company, Research and Development, Tokyo, and Department of Gastroenterology, Graduate School of Medicine, University of Tokyo, Tokyo, Japan
ABSTRACT
Background and Aim: Entecavir has demonstrated clinical efficacy for chronic hepatitis B. This study evaluated the efficacy and safety of entecavir in nucleoside-naive Japanese chronic hepatitis B patients.
Methods: In this multicenter, double-blind study, 66 nucleoside-naive Japanese chronic hepatitis B patients were randomized to 0.1 mg entecavir (n = 32) or 0.5 mg entecavir (n = 34) daily for 52 weeks. The primary endpoint was the proportion of patients whose serum hepatitis B virus (HBV) DNA decreased from baseline by ≥2 log10 copies/mL or became undetectable (<400 copies/mL by polymerase chain reaction assay) at week 48.
Results: One hundred percent of patients in both treatment groups achieved the primary efficacy endpoint, with 81% and 68% of patients achieving undetectable HBV DNA in the 0.1 mg and 0.5 mg treatment groups, respectively. Mean changes from baseline in HBV DNA were -4.49 log10 and -4.84 log10 copies/mL for the 0.1 mg and 0.5 mg groups, respectively. Significant improvements in necroinflammation were seen in both groups, as assessed by Knodell and New Inuyama classifications. Most adverse events were transient and classified as grade 1 or 2. There were no clinically significant differences in adverse events across the two treatment groups and no discontinuations due to adverse events in either group.
Conclusions: In Japanese nucleoside-naive patients with chronic hepatitis B, 0.1 mg or 0.5 mg entecavir daily provided excellent efficacy and was well tolerated. The 0.5 mg dose was selected for the treatment of nucleoside-naive patients.
Introduction
It has been reported that 350-400 million people worldwide are chronically infected with hepatitis B virus (HBV)1,2 despite the widespread use of HBV vaccination for prevention of this disease. HBV infection is particularly prevalent in Asia-Pacific countries, with an estimated 75% of all chronically infected patients living in the region.3 Prevalence rates reported in 2000 indicated that 0.8% of the Japanese population were hepatitis B virus surface antigen (HBsAg) positive with 36% of infected individuals being chronically infected.4 Among those chronically infected, 20-40% will develop cirrhosis, decompensated liver disease or hepatocellular carcinoma.5 In Asia, HBV is the leading cause of chronic hepatitis, cirrhosis and hepatocellular carcinoma.4
Treatment for chronic hepatitis B has evolved markedly over the last decade. Interferon-_ was the only available treatment for many years, but this cytokine is efficacious in only approximately 35% of patients,6 and is poorly tolerated by many patients due to adverse effects. Lamivudine, a cytosine analog, was the first oral anti-HBV nucleoside analog developed, and has demonstrated efficacy for treatment of chronic hepatitis B during short-term administration.7,8 However, viral breakthrough due to emergence of lamivudine-resistant strains of HBV with amino acid substitutions in the YMDD (tyrosine-methionine-aspartate-aspartate) motif of reverse transcriptase result in loss of clinical benefit.9-11 Long-term follow up of hepatitis B e antigen (HBeAg)-negative patients treated with adefovir, the second approved oral anti-HBV, have shown cumulative probabilities of genotypic resistance of 29% at 5 years12 and some studies have reported that 20-50% of patients receiving a 10 mg dose of adefovir have primary non-response13 indicating that the approved dose of adefovir may be suboptimal.14
Entecavir is a deoxyguanosine analog that has more than 300 times greater potency than lamivudine in vitro.15,16 Entecavir inhibits all three steps of HBV DNA replication: (i) priming of the HBV DNA polymerase; (ii) reverse transcription of negative-strand HBV DNA from pre-genomic messenger RNA; and (iii) synthesis of positive-strand HBV DNA.17 In woodchuck models of HBV infection, entecavir reduced viral loads by up to 9 log10 copies/mL and prevented the onset of hepatocellular carcinoma.18 In early clinical studies, entecavir was demonstrated to be safe and efficacious when given for 28 days.19 In a 24-week, phase II international clinical trial, Lai et al. demonstrated a dose-response relationship for entecavir, and showed that entecavir was superior to lamivudine at doses of 0.1 mg and 0.5 mg for HBV DNA reduction.20 Subsequently, two phase III international trials showed that 0.5 mg entecavir daily for 48 weeks achieved superior histological, virological and biochemical improvement in HBeAg-positive and HBeAg-negative nucleoside-naive patients compared with lamivudine, with comparable safety and no emergence of resistance without prior existence of the amino acid substitutions rtL180M and rtM204V/I/S which are associated with lamivudine resistance.21,22 Entecavir was approved by the US regulatory authorities in March 2005. Study AI463047 evaluated 0.01 mg, 0.1 mg and 0.5 mg entecavir and 100 mg lamivudine in nucleoside-naive Japanese patients and established the 0.5 mg dose of entecavir as the optimal dose in this patient population. The current phase II dose-ranging trial, which commenced before the completion of AI463047, evaluated the efficacy and safety of 0.1 mg and 0.5 mg entecavir daily for 52 weeks in nucleoside-naive chronic hepatitis B patients in Japan. This study's primary objective was to demonstrate that entecavir has antiviral activity as indicated by the proportion of subjects who achieve a reduction from baseline in HBV DNA by ≥2 log10 copies/mL or to <400 copies/ml at week 48.
Methods
Study design
This was a randomized, double-blind, multicenter trial of 0.1 mg entecavir once daily and 0.5 mg entecavir once daily for 52 weeks in nucleoside-naive patients with HBeAg-positive or -negative chronic hepatitis B. Patients were randomized via a central registration procedure. A total of 66 patients were enrolled in this study, including men and women ranging in age from 27-68 years who were determined to be eligible for the study during the 6-week screening period. Following randomization, patients received either a 0.1 mg entecavir tablet plus a 0.5 mg placebo tablet (n = 32) or a 0.5 mg entecavir tablet plus a 0.1 mg placebo tablet (n = 34) orally once daily for 52 weeks. After 52 weeks of blinded dosing, patients were given the option of enrolling in an entecavir rollover study. All patients who discontinued blinded dosing early, or who completed the protocol but did not enroll in the entecavir rollover study, were followed for 24 weeks post-dosing, and could receive marketed anti-HBV therapy as recommended by their physician.
The study was conducted in compliance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and Articles/Notifications of the Ministry of Health, Labor and Welfare in Japan. Written informed consent was obtained from all patients.
The study's primary efficacy objective was to demonstrate that 0.1 mg and 0.5 mg doses of entecavir had antiviral activity as indicated by the proportion of patients who achieve a reduction in HBV DNA of ≥2 log10 copies/mL or to below the limit of quantification (LOQ, 400 copies/mL) by polymerase chain reaction (PCR) assay (Roche Amplicor, Hoffmann-La Roche Ltd, Basel, Switzerland) at week 48. Secondary endpoints included the mean change from baseline in HBV DNA by PCR assay at week 48, and proportions of patients who achieved the following at week 48: (i) HBV DNA less than 400 copies/mL; (ii) serum alanine aminotransferase (ALT) normalization (<1.25 times the upper limit of normal [ULN], World Health Organization [WHO] toxicity grade 0); (iii) HBeAg loss and HBeAg seroconversion (HBeAg loss and appearance of anti-HBe) among patients who were HBeAg-positive at baseline; and (iv) complete response, defined as HBV DNA less than 400 copies/mL by PCR assay plus ALT less than 1.25 X ULN plus HBeAg negativity for those who were HBeAg-positive at baseline. The incidence of amino acid substitutions associated with entecavir resistance in patients who experienced a virological breakthrough, defined as an increase in HBV DNA of ≥1 log10 copies/mL from nadir, was also determined. Among patients with evaluable baseline and week 48 liver biopsies, the proportion of patients with histological improvement was determined. Histological improvement was defined as a ≥2-point decrease in the Knodell necroinflammatory score and no worsening of fibrosis (worsening was defined as a ≥1-point increase in the Knodell fibrosis score) from baseline to week 48. Liver biopsies were also evaluated using the New Inuyama classification system. The biopsy reading committee was blinded to treatment and sequence.
The primary safety endpoint was the proportion of patients in each treatment group who discontinued study medication due to adverse events. Secondary safety endpoints included incidence of adverse events, serious adverse events, laboratory abnormalities, grade 3-4 clinical adverse events and grade 3-4 laboratory abnormalities. ALT flares were defined as ALT >2 x baseline and >10 X ULN.
Study population
Patients were eligible for enrollment if they met the following inclusion criteria: (i) hepatitis B surface antigen (HBsAg)-positive for 24 weeks or more prior to screening or HBsAg-positive for less than 24 weeks prior to screening, negative for immunoglobulin M anti-hepatitis B core antibody and confirmation of chronic hepatitis on liver biopsy; (ii) HBeAg-positive for more than 12 weeks prior to screening or HBeAg-negative and positive for anti-HBe; (iii) active viral replication as evidenced by HBV DNA of ≥105 copies/mL by PCR assay at screening; (iv) serum ALT ranging 1.3-10 times the ULN; and (v) compensated liver disease, as indicated by a prothrombin time ≦3 s longer than the normal control value or international normalized ratio of ≦1.5, serum albumin of ≥3.0 g/dL (30 g/L) and total bilirubin of ≦2.5 mg/dL (42.75 μmol/L). Women of childbearing potential underwent contraceptive procedures as appropriate to avoid pregnancy during the trial period and for up to 8 weeks after completion of the trial.
The following patients were excluded from the study: pregnant and nursing women; patients diagnosed with cirrhosis, or with a history or evidence of variceal bleeding, encephalopathy or ascites requiring diuretics or paracentesis; patients with other forms of liver disease or suspected hepatic tumors; patients diagnosed with HIV infection; patients with a history of pancreatitis within 24 weeks prior to initiation of protocol therapy; and patients with an increased risk of hepatic toxicity or pancreatitis. In addition, patients who received immunosuppressive therapy (including systemic administration of corticosteroid-derivative agents) or who were treated with interferon-_ or -_, within 24 weeks prior to initiation of protocol therapy, were excluded from the study. Patients treated with anti-HBV nucleoside analogs for more than 12 weeks were also excluded.
Assay methods
Serum HBV DNA was determined by Roche Amplicor PCR assay (LOQ, 400 copies/mL; Roche Diagnostics, Tokyo, Japan)23 in a central laboratory. Clinical laboratory tests, PCR assays for HBV DNA, and serological tests for HBV were performed at SRL Inc. (Tokyo, Japan), the central clinical laboratory designated by the trial sponsor. Liver biopsy was performed within 6 weeks of initiation of study therapy; or, if a liver biopsy had been previously obtained within 52 weeks before initiation of protocol therapy, it was used as the baseline specimen for histological evaluation. Baseline biopsies were evaluated using the Knodell Histological Activity Index (HAI) and Knodell fibrosis scores,24 and the New Inuyama classifications.25 Genotype analysis of HBV strains was performed on samples from all patients at baseline using a PCR-restriction fragment length polymorphism assay (SRL). All samples were also analyzed at baseline for evidence of amino acid substitutions associated with lamivudine resistance (rtM204V/I) using a PCR-enzyme-linked minisequence assay (Medical & Biological Laboratories, Aichi, Japan). In addition, patients who experienced virological breakthrough (increase in HBV DNA of ≥1 log10 copies/mL from nadir of treatment) had baseline and on-treatment samples analyzed for amino acid substitutions associated with entecavir resistance (rtT184, rtS202 and rtM250) using a HBV DNA polymerase sequence assay at SRL.
Statistical analysis
Analyses of efficacy endpoints were based on treated patients. The primary objective would be demonstrated if the lower limit of the 95% confidence interval for the proportion of patients who achieved a reduction in HBV DNA from baseline by ≥2 log10 copies/mL or to less than 400 copies/mL by PCR assay at week 48 in either treatment arm was at least 60%. Parameters represented by continuous variables were summarized by the mean and standard error. In the analysis of binary endpoints, patients with missing week 48 measurements were treated as missing (non-completer = missing). All reported P-values are two-sided. For comparison of liver biopsy specimens before and after treatment, a Wilcoxin signed-rank test was performed.
Results
Study population
Of 102 patients enrolled and screened, 66 were randomized and treated. Thirty-two patients were assigned 0.1 mg entecavir and 34 patients 0.5 mg entecavir. The two treatment groups were well balanced at baseline for demographic and disease-related characteristics (Table 1). Approximately 80% of patients in both groups were HBeAg-positive, and mean HBV DNA at baseline were 7.26 and 7.68 log10 copies/mL for the 0.1 mg and 0.5 mg groups, respectively. Overall, 62 patients were infected with HBV genotype C, and two patients were infected with HBV genotype B. In two patients, HBV genotype was not identified. All patients completed protocol therapy for 52 weeks and were assessed for efficacy and safety. Compliance, measured by the volume of unused product returned from subjects to the institution, was reported to be between 95% and 100%. After completion of the protocol therapy, all patients entered an entecavir rollover study.
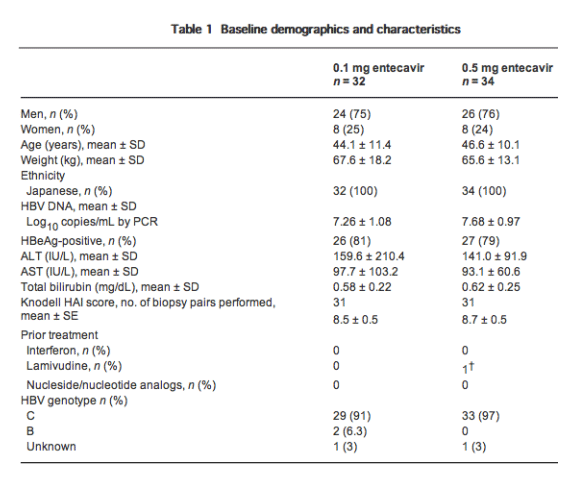
Virological response
One hundred percent of patients in both treatment groups achieved the primary efficacy endpoint (a reduction from baseline in HBV DNA of ≥2 log10 copies/mL or to <400 copies/mL by PCR assay at week 48; Table 2). By week 4, 94% and 91% of patients in the 0.1 mg and 0.5 mg groups, respectively, achieved the primary endpoint; 100% of patients in both groups had achieved it by week 8 and that proportion was maintained through to the end of treatment (week 48). Mean serum HBV DNA declined rapidly in both groups through week 4, and thereafter declined more slowly (Fig. 1). Mean change from baseline in HBV DNA at week 24 was -4.43 log10 copies/mL for the 0.1 mg group and -4.79 log10 copies/mL for the 0.5 mg group. Between weeks 24 and 48, across both groups, only slight decreases in mean HBV DNA occurred. At week 48, mean change from baseline in HBV DNA was -4.49 log10 copies/mL for the 0.1 mg group and -4.84 log10 copies/mL for the 0.5 mg group (P = non-significant [NS]; Fig. 1, Table 2). Eighty-one percent of patients receiving 0.1 mg entecavir and 68% of patients receiving 0.5 mg entecavir achieved HBV DNA of less than 400 copies/mL by PCR assay at week 48 (P = NS, Table 2).
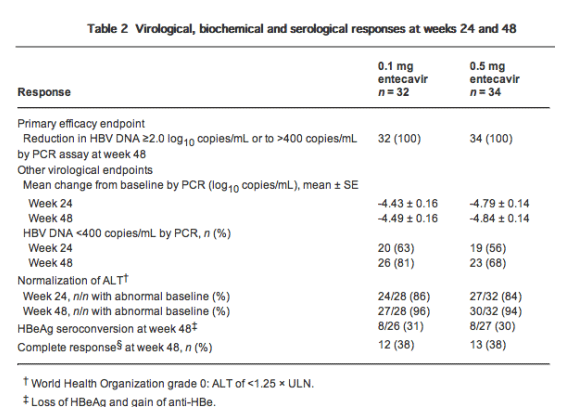
Figure 1 Mean change from baseline in hepatitis B virus (HBV) DNA through week 48 by polymerase chain reaction assay (log10 copies/mL) in patients treated with 0.1 mg and 0.5 mg entecavir (ETV). Data expressed as mean ± standard error.
Biochemical response
Approximately 90% of patients demonstrated abnormal ALT (≥1.25 X ULN) at baseline. Among patients with abnormal baseline ALT, the proportions achieving ALT normalization (<1.25 X ULN; WHO toxicity grade 0) at week 48 were 96% (27/28) for patients receiving 0.1 mg entecavir and 94% (30/32) for patients receiving 0.5 mg entecavir (P = NS, Table 2).
Serological response
Among patients who were HBeAg-positive at baseline, the proportions achieving HBeAg loss at week 48 were 31% (8/26) in the 0.1 mg group and 30% (8/27) in the 0.5 mg group (Table 2). All patients who demonstrated HBeAg loss also showed acquisition of anti-HBe, thus rates of HBeAg seroconversion at week 48 were also 31% and 30% for the 0.1 mg group and 0.5 mg groups, respectively (Table 2).
Complete response
At week 48, the proportions of patients achieving complete response (defined as HBV DNA <400 copies/mL by PCR assay plus ALT <1.25 X ULN plus HBeAg negativity if they were HBeAg-positive at baseline) were 38% (12/32) for patients receiving 0.1 mg entecavir and 38% (13/34) for patients receiving 0.5 mg entecavir (P = NS, Table 2).
Histological response
Ninety-one percent (29/32) of patients in the 0.1 mg entecavir group and 88% (30/34) of patients in the 0.5 mg entecavir group had evaluable biopsy pairs from baseline and week 48 (Table 3). Histological improvement, defined using the Knodell classification system, occurred in 72% (21/29) and 80% (24/30) of patients in the 0.1 mg and the 0.5 mg groups, respectively. Mean change in Knodell HAI scores were _3.2 and -4.6 for the 0.1 mg and the 0.5 mg groups, respectively. For both groups, the change from baseline in HAI score was significant (P < 0.0001 for both groups). In patients who received 0.5 mg entecavir, 29% (9/31) of patients experienced an improvement or no worsening of Knodell fibrosis score, and the mean change from baseline in Knodell fibrosis score from baseline was significant (P = 0.004). According to New Inuyama classification, grading of necrotic/inflammatory findings improved for 64% (20/31) of patients in the 0.1 mg group and 74% (23/31) of patients in the 0.5 mg group, while no patient demonstrated worsening. For both groups, the improvement from baseline was significant (P < 0.0001 for both comparisons) (Table 3). According to the New Inuyama fibrosis staging system, improvement in fibrosis occurred in 24% (7/29) of patients in the 0.1 mg group (P = NS) and in 40% (12/30) of patients in the 0.5 mg group (P = 0.003, Table 3).
Resistance analysis
During the treatment period, two patients who received 0.5 mg entecavir experienced virological breakthrough at week 36. Both patients achieved undetectable HBV DNA. The first patient, with a baseline HBV DNA of 8.4 log10 copies/mL, experienced an increase in HBV DNA to 3.6 log10 copies/mL which was maintained at 48 weeks. The second patient, with a baseline HBV DNA of 8.5 log10 copies/mL, experienced an increase in HBV DNA to 4.5 log10 copies/mL and a subsequent decrease to 3.1 log10 copies/mL at week 48. Neither of the patients experienced ALT flares or other clinically relevant events. Genotypic analysis of HBV DNA polymerase was performed on samples from these two patients. Neither L180M nor M204V/I/S (which are associated with lamivudine resistance)17,26,27 nor amino acid substitutions associated with entecavir resistance (at positions T184, S202 and M250) were detected.28 Genotypic analysis of virus from all patients was carried out at baseline using methods with a sensitivity cut-off of 25%. Neither rtL180M nor rtM204V/I/S was detected in any patient. At week 48, the amino acid substitution rtM204I (associated with lamivudine resistance) was detected in two patients, one in the 0.1 mg group and one in the 0.5 mg group. However, entecavir was efficacious in the presence of the rtM204I variants and neither patient demonstrated virological breakthrough (HBV DNA decreased by 3.1 log10 copies/mL in the patient in the 0.1 mg group, and reduced to <2.6 log10 copies/mL in one patient in the 0.5 mg group at week 48) or elevation of ALT.
Safety
All 66 patients treated with the study drug completed 52 weeks of dosing. Adverse events were reported for all patients, but most were transient and mild or moderate (grade 1-2) in severity (Table 4). Adverse events were not considered to be related to the study drug, except for a number of cases of headache (five in the 0.1 mg arm and eight in the 0.5 mg arm), all of which were grade 1 or 2. Grade 3-4 clinical adverse events were observed in two patients (6%) in each treatment group, none of which was related to the study drug (one case of enteritis and one of spondylolisthesis in the 0.1 mg arm; one case of enteritis and one of intervertebral disc herniation in the 0.5 mg arm). Grade 3-4 laboratory adverse events occurred in five (16%) and six (18%) of patients in the 0.1 mg and 0.5 mg groups, respectively (two cases of ALT and AST elevations, two of lipase elevations and one of blood glucose elevation in the 0.1 mg arm; two cases of ALT and AST elevations, one of ALT elevation, one of lipase elevation, one of blood glucose elevation and one of amylase elevation in the 0.5 mg arm). There were no deaths in the study.
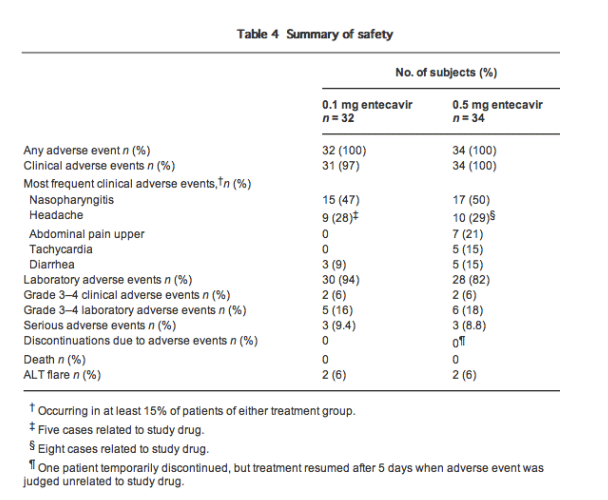
Serious adverse events occurred in three (9%) of patients in each treatment group (one case of infectious enterocolitis, one of acquired spondylolisthesis and one of ALT elevation in the 0.1 mg arm; one case of duodenal ulcer hemorrhage, one of ligament damage and one of intervertebral disc protrusion in the 0.5 mg arm). No serious adverse event was judged by the investigator to be related to study medication. The protocol therapy was temporarily discontinued for one patient who developed duodenal ulcer hemorrhage, but therapy was restarted after 5 days when causal relationship with the test medication was ruled out. Except for this temporary interruption, no patient discontinued therapy for adverse events.
Alanine aminotransferase flares (defined as ALT of >2 times baseline and of >10 X ULN: grade 4) occurred in two patients (6%) in the 0.1 mg entecavir group and two patients (6%) in the 0.5 mg entecavir group. All ALT flares were transient, resolved on treatment, and were associated with a ≥2 log10 copies/mL reduction in HBV DNA. No ALT flare was associated with signs or symptoms of hepatic decompensation.
Discussion
The current study demonstrates that entecavir dosing for 52 weeks in Japanese patients was highly effective in reducing HBV DNA and normalizing ALT. The primary objective of this study was to demonstrate that entecavir has antiviral activity as indicated by the proportion of subjects who achieve a reduction from baseline in HBV DNA by ≥2 log10 copies/mL or to <400 copies/mL at week 48. All patients in both treatment groups achieved this primary efficacy endpoint, underscoring entecavir's potent anti-HBV efficacy. In both treatment groups, serum HBV DNA declined rapidly through week 4, then declined more slowly through week 24, and continued to decline through week 48 (mean change from baseline of -4.49 ± 0.16 and -4.84 ± 0.14 log10 copies/mL for 0.1 mg and 0.5 mg entecavir, respectively). This profile confirms the typical multiphasic pattern of antiviral action against HBV, similar to that observed in an international phase II 24-week trial of entecavir.20
Entecavir treatment also resulted in ALT normalization (ALT of <1.25 X ULN: WHO grade 0 toxicity) and HBeAg seroconversion concurrent with the observed declines in HBV DNA. Overall (across both treatment groups), approximately 95% of patients achieved ALT normalization, and approximately 30% of HBeAg-positive patients achieved HBeAg seroconversion. These results are similar to those reported in other international clinical trials of entecavir.21,22 Specifically, Chang et al. reported that 48 weeks of entecavir 0.5 mg daily in nucleoside-naive, HBeAg-positive patients resulted in ALT normalization (ALT of ≦1.0 X ULN) in 68% and HBeAg seroconversion in 21% of these patients.21 In the same study, 67% of patients achieved undetectable HBV DNA at week 48, which is comparable to the results of the present study. However, the mean change from baseline in HBV DNA in the study by Chang et al. (-6.9 log10 copies/mL) was greater than was observed in the current study (-4.84 log10 copies/mL for the entecavir 0.5 mg group). This difference is accounted for by the higher baseline HBV DNA of patients in the international trial (9.6 log10 copies/mL vs 7.7 log10 copies/mL for the 0.5 mg group in the present study).
The ultimate goal of chronic hepatitis B treatment is to arrest or reverse liver disease progression associated with HBV infection. This parameter is most directly and reliably measured by histological evaluation. In the current study, entecavir at doses of both 0.1 mg and 0.5 mg daily resulted in high rates of histological improvement (72% and 80%, respectively) when assessed by the Knodell scoring system. As the New Inuyama classification system is most often used to grade and stage liver disease progression in Japan, we also employed this method of histological evaluation, and obtained results consistent with the results of the Knodell evaluations; that is, there was significant improvement in necrosis/inflammation across both entecavir treatment groups and significant improvement in fibrosis for the 0.5 mg entecavir group. Entecavir's demonstrated histological benefit after 1 year of treatment suggests that its potent viral suppression might also reduce the risk of progression to cirrhosis and end-stage liver disease among chronic hepatitis B patients.
A high barrier to resistance among nucleoside-naive patients has been demonstrated with entecavir.28,29 The combination of potent viral suppression and the requirement for multiple amino acid substitutions in the HBV reverse transcriptase to confer resistance to entecavir suggests that resistance emergence will be a rare event during long-term administration of entecavir. In phase III international clinical trials, less than 1% of patients treated with entecavir through 2 years experienced a virological breakthrough due to the emergence of entecavir resistance.29 Phenotypic analyses have demonstrated that entecavir-resistant strains do not emerge in the absence of amino acid substitutions associated with lamivudine resistance (rtL180M and/or rtM204V/I/S).29 In the present study, no amino acid substitutions at T184, S202 or M250 (all of which can mediate resistance in the presence of rtL180M + rtM204V/I/S) were detected. rtM204I emerged in two patients, one in the 0.1 mg entecavir group and one in the 0.5 mg entecavir group. In both patients, entecavir continued to suppress HBV replication and virological breakthroughs were not observed.
Entecavir was generally well tolerated in the current study. There were no clinically significant differences in the incidence or severity of adverse events between the two treatment groups, indicating that entecavir was well tolerated at a daily dose of 0.5 mg. ALT flares were infrequent, and those flares that did occur were associated with reductions in HBV DNA, and resolved without treatment interruption. These results are consistent with the safety and tolerability profile of entecavir reported in international trials.20-22
Entecavir's potent antiviral efficacy, good tolerability and high barrier to resistance offer the potential for long-term treatment of chronic hepatitis B with the objective of halting or reversing liver disease progression. The mean reduction in HBV DNA from baseline at week 48 and histological improvement observed in this trial, together with the results of previously published international trials, support the selection of the 0.5 mg dose of entecavir as an appropriate choice of primary therapy for treatment of nucleoside-naive Japanese patients with chronic hepatitis B infection.
|
|
| |
| |
|
|
|