| |
Hepatitis B Virus Resistance to Nucleos(t)ide Analogues
|
| |
| |
Gastroenterology Nov 2009
Fabien ZoulimCorresponding Author Informationemail address, Stephen Locarnini
INSERM, U871, Lyon, France
Universite Lyon 1, Lyon, France
Hospices Civils de Lyon, Service d'hepatologie et de gastroenterologie, Lyon, France
Victorian Infectious Diseases Reference Laboratory, North Melbourne, Victoria, Australia
"best strategies for preventing NA resistance include first-line use of the most potent antivirals with a high barrier to resistance....The spread of drug-resistant HBV mutants can be reduced by avoiding unnecessary drug use, choosing drugs and combinations more carefully, and continually monitoring or carrying out targeted surveillance for drug resistance"
"The choice of the first-line agent(s) is very important; the goal is to delay the development of resistance and to preserve treatment options over the long-term of treatment for CHB. The development of therapeutics that complement the effects of NA is warranted; new treatment strategies must be aimed at increasing viral suppression and promoting virologic clearance and ensuring the prevention of drug resistance and its complications."
Patients with chronic hepatitis B (CHB) can be successfully treated using nucleos(t)ide analogs (NA), but drug-resistant hepatitis B virus (HBV) mutants frequently arise, leading to treatment failure and progression to liver disease. There has been much research into the mechanisms of resistance to NA and selection of these mutants. Five NA have been approved by the US Food and Drug Administration for treatment of CHB; it is unlikely that any more NA will be developed in the near future, so it is important to better understand mechanisms of cross-resistance (when a mutation that mediates resistance to one NA also confers resistance to another) and design more effective therapeutic strategies for these 5 agents. The genes that encode the polymerase and envelope proteins of HBV overlap, so resistance mutations in polymerase usually affect the hepatitis B surface antigen; these alterations affect infectivity, vaccine efficacy, pathogenesis of liver disease, and transmission throughout the population. Associations between HBV genotype and resistance phenotype have allowed cross-resistance profiles to be determined for many commonly detected mutants, so genotyping assays can be used to adapt therapy. Patients that experience virologic breakthrough or partial response to their primary therapy can often be successfully treated with a second NA, if this drug is given at early stages of these events. However, best strategies for preventing NA resistance include first-line use of the most potent antivirals with a high barrier to resistance. It is important to continue basic research into HBV replication and pathogenic mechanisms to identify new therapeutic targets, develop novel antiviral agents, design combination therapies that prevent drug resistance, and decrease the incidence of complications of CHB.
The hepatitis B virus (HBV) is a DNA virus that replicates its genome via an RNA intermediate using reverse transcription1 (Supplementary Figure 1); chronic infection with this virus can result in cirrhosis and hepatocellular carcinoma.2 Effective treatments have been developed for chronic hepatitis B (CHB), significantly reducing morbidity and mortality. Therapeutic efficacy can be affected by factors such as the development of adverse effects, poor patient compliance, previous treatment with suboptimal regimens, infection with drug-resistant viral strains, and inadequate drug exposure because of pharmacologic properties of particular drug(s) and individual genetic variation. Interferon (conventional or pegylated) and 5 other drugs that belong to the class of nucleos(t)ide analogues (NA) (lamivudine [LMV], adefovir dipivoxil [ADV], entecavir [ETV], telbivudine [LdT], and tenofovir [TDF]) have been approved for treatment of CHB in many parts of the world.2 The NA inhibit reverse transcription of the HBV polymerase, and this article reviews the emergence of resistance to this class of agents.
Clinical Importance of Resistance
Impact on Disease Severity
The development of drug resistance begins with mutations in the polymerase gene, followed by an increase in viral load, an increase in serum alanine aminotransferase (ALT) levels several weeks to months later, and progression of liver disease3, 4, 5 (Figure 1). In patients with LMV resistance, the risk of increased serum ALT is usually correlated with the duration of infection with the mutant virus.6 These patients are also at significant risk of ALT flare, which may be accompanied by hepatic decompensation.6 The emergence of LMV-resistant mutations is reflected in histologic assessment of liver samples.7 The detrimental effect of HBV drug resistance on clinical outcome was shown by a placebo-controlled trial of LMV in patients with advanced fibrosis.8 Patients successfully treated with LMV who maintained wild-type HBV had a significantly lower risk of liver disease progression compared with those who received placebo, but this effect was lost in patients who developed LMV-resistant mutant forms of the virus.8
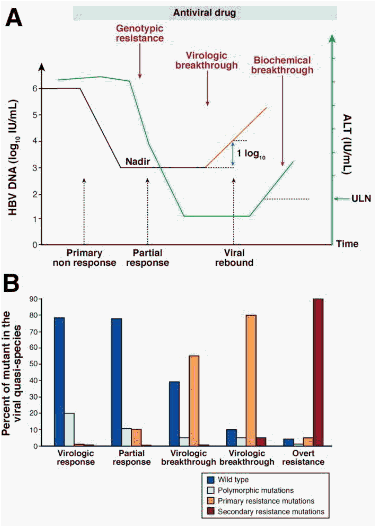
Figure 1. Kinetics of drug resistance emergence. Panel A: Evolution of viral load and ALT levels. After an initial drop in viral load following the initiation of antiviral therapy, virologic breakthrough may occur as a consequence of antiviral drug resistance. It corresponds to the rise in serum HBV DNA levels of at least 1-log10 IU/mL compared with the lowest value during therapy (nadir value), in 2 consecutive samples 1 month apart, in patients who have previously responded and have a good treatment compliance. It may be followed by an elevation in serum ALT levels in patients who previously showed transaminases normalization under treatment. It may result in hepatitis flares and in worsening of liver histology. Panel B: Evolution of the viral quasispecies with respect to primary and secondary resistance mutations. At the beginning of therapy, wild-type virus is the major strain circulating in the patient's blood, whereas viral genomes harboring polymorphic mutations may be detected. Because of the spontaneous error rate of the viral polymerase, primary resistance mutations are usually present at levels that are undetectable by conventional diagnostic techniques. At the time of virologic breakthrough, viral genomes harboring primary resistance mutations start to emerge and become the dominant viral strains. The continuation of viral replication under the selective pressure of the drug may lead to the accumulation of additional mutations that increase the resistant mutant replication capacity (ie, secondary resistance mutations).
The kinetics of emergence of resistance to ADV are typically slower than those of LMV but follow the same sequence of events: polymerase variants with the specific resistance mutations can be detected initially, which is next followed by virologic breakthrough and then rising serum levels of ALT.9 In some cases, the emergence of ADV resistance was also associated with acute exacerbation of disease and liver failure.10
Only limited data are available on the clinical outcome of patients who are infected with LdT-, ETV-, or TDF-resistant HBV, mainly because treatment adaptation, usually based on in vitro cross-resistance data, has been initiated during early stages of resistance, or treatment failure. Thus, the availability of antiviral drugs with complementary cross-resistance profiles (see below) has changed the management of patients with drug resistance, allowing physicians to prevent the worsening of clinical outcome resulting from the emergence of resistance.
Incidence and Prevalence of Resistance
LMV resistance increases progressively over the course of treatment; 14%-32% of patients become resistant to the drug each year after treatment was initiated, and more than 80% are resistant after 48 months of treatment5 (Table 1). The rate of emergence of LdT-resistant HBV is lower than that of LMV but is still substantial. In a phase III trial that compared LdT with LMV, genotypic resistance occurred in 4.4% and 21% of hepatitis B e antigen (HBeAg)-positive patients after 1 and 2 years of treatment, respectively, and 2.7% and 8.6% in HBeAg-negative patients, respectively11, 12 (Table 1)
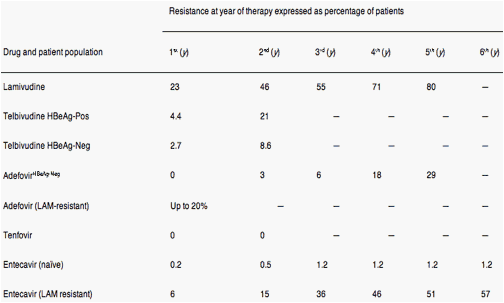
The rate of selection for ADV-resistant virus is lower; resistance occurs in approximately 2% of patients with HBeAg-negative CHB after 2 years of therapy (Table 1). However, following 4-5 years of ADV monotherapy, up to 30% of patients are found to be resistant.9, 13 When ADV has been used in patients who are resistant to LMV, primary ADV resistance as detected by genotype analysis has been found in up to 20% of patients by 12 months after ADV therapy began.14 Recent trials of TDF reported that no resistance had developed by weeks 4815 and 96 of treatment, although at week 72, the majority of viremic patients was given a combination of TDF and emtricitabine (Truvada; Gilead Sciences, Inc, Foster City, CA).16
Very low rates of genotypic resistance to ETV have been reported in treatment-naïve patients after 1 year (0.1%), 2 years (0.4%), 3 years (1.2%), 4 years (1.2%), 5 years (1.2%), and 6 years (1.2%) of therapy17, 18 (Table 1). In contrast, in patients previously treated with LMV, the cumulative genotypic resistance rates are 6% (year 1), 14% (year 2), and 32% (year 3) (Table 1), steadily increasing to almost 60% by year 6.17, 18
Principles of Resistance and Cross-resistance
A major determinant in the slow kinetics of HBV clearance from infected cells is the presence of a replicative form of the viral DNA termed covalently closed circular DNA (cccDNA)19, 20 (Supplementary Figure 1). During chronic HBV infection, cccDNA is maintained in the hepatocyte nuclei with a long half-life in infected cells.21 Furthermore, it has been shown that antiviral therapy with NA cannot prevent the initial formation of cccDNA, indicating that persistent viremia during therapy leads to infection of new cells.22, 23 The HBV cccDNA acts as a reservoir for the reactivation of viral genome replication and is responsible for viral relapse after withdrawal of antiviral therapy or those patients with CHB with immune suppression. It has also been shown in the woodchuck model of hepadnavirus infection that drug resistance mutations are archived in cccDNA and may therefore be selected rapidly out when using drugs that exhibit cross-resistance.24 Thus, the stability and replenishment of cccDNA is the stumbling block for eradicating CHB infection with current antiviral agents.25, 26, 27, 28
A combination of host and viral factors determine viral persistence and also NA resistance. Infected hepatocytes have a long half-life, contributing to HBV persistence in the liver.29, 30, 31, 32 Mathematical modeling showed that the half-life of hepatocytes varies from 30 to 100 days, depending on individuals' immune response.33, 34 HBV genome variability during the chronic phase of the disease determines the selection for viral resistant strains.20, 35, 36 Clonal and pyrosequencing analysis of HBV genomes have shown that single mutants exist in the overall viral population of HBVs even before therapy begins. Viral quasispecies within the same patient evolve during the course of infection: different variants or mutants are selected at different stages of infection in response to the host immune response or antiviral therapy (Figure 2).
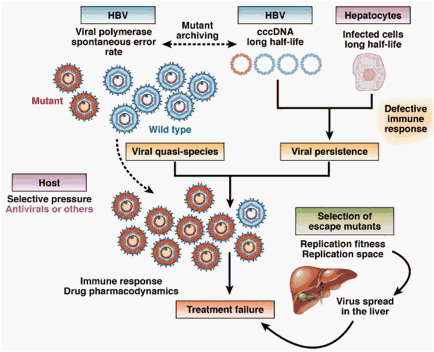
Figure 2. Mechanisms of selection and emergence of HBV drug-resistant mutants. The main factors involved in the selection of escape mutants are: (i) the long half-life of hepatocytes and viral cccDNA; (ii) the HBV genome variability leading to a complex viral quasispecies and mutant archiving in cccDNA. The composition of the viral quasispecies evolves over time depending on the selective pressure including antiviral therapy and the host immune response. Escape mutants may then spread in the liver and become the dominant species depending on their fitness (ie, their capacity to replicate and dominate wild-type strain in the presence of antiviral pressure) and the replication space available for their dissemination in the liver. Their selection results in treatment failure.
Different mechanisms are involved in the selection of drug-resistant mutants during antiviral therapy.20, 35 As described above, a complex mixture of genetically distinct variants have a replicative advantage in the presence of the selective pressure of NA therapy. A newly acquired or a preexisting mutation conferring a selective advantage to a variant will generate progeny virus, which is more fit and can spread more rapidly in the liver, allowing the corresponding mutant to accumulate and become the dominant species in the liver in the presence of the antiviral drug.37, 38, 39, 40 The replacement of wild-type virus in liver cells by a dominant mutant is a slow process; studies in animal models indicate that resistant mutants predominately infect uninfected cells (ie, replication space), so the spread of the dominant mutant depends on the number of uninfected cells in which HBV can replicate.24, 41 It might take months for the immune system to remove hepatocytes that are infected with wild-type HBV and for new hepatocytes to develop that are susceptible to infection by viral drug-resistant mutant HBV. The infectivity of the drug-resistant mutants can impact the speed of their selection; mutations in the genes that encode overlapping surface antigens can affect viral fitness and infectivity,42 virion release (because of intracellular retention of newly synthesized virus),43 virologic breakthrough (with slower kinetics of viral load increase),43 and vaccine prophylaxis.44 Finally, the level of resistance to a drug, usually conferred by specific mutation in the viral polymerase, not surprisingly affects the fitness of the mutant.
Antiviral drug resistance results from adaptive mutations in the viral genome.45 HBV infection is characterized by very high levels of virus production and turnover, producing more than 1011 virions per day.46 Furthermore, the viral population in an infected person is highly heterogeneous.47 The high rate of HBV replication, combined with the high mutation rate (1 in every 105 nucleotide substitutions during each cycle of replication, because of the error-prone nature of reverse transcription48), results in patients with CHB having a diverse mixture of viral quasispecies, each differing in 1 or more mutations.49 The probability that a mutation associated with drug resistance is selected for during therapy also depends on the potency of that drug.50 Replication fitness (defined as the ability to replicate under selection pressure45) and the replication capacity of resistant isolates can shape the pattern of primary vs secondary mutations that emerge.51 The availability of replication space for HBV also determines resistance; the liver can accommodate new transcriptional templates of cccDNA only if uninfected cells are generated by normal liver growth or hepatocyte proliferation or by direct loss of cccDNA.52 The genetic barrier to resistance of the treatment regimen increases as the number of specific mutations required for drug resistance increases.53 Finally, antiviral drug resistance is also affected by host characteristics of virus-infected hepatocytes, immune response, and genetic background.
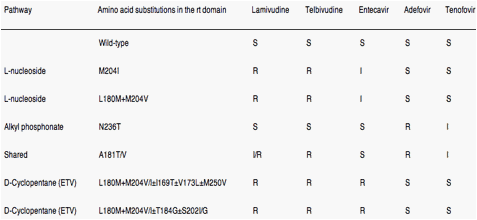
Cross-resistance is defined as resistance to drug(s) to which a virus has never been exposed. From a cross-resistance perspective, the 5 approved NA have been placed, based on structural characteristics, into 3 groups: L-nucleosides (LMV and LdT), alkyl phosphonates (ADV and TDF), and D-cyclopentane group (ETV). Resistance and cross-resistance tend to be structure specific.
Two types of mutations have been associated with treatment failure to NA: primary resistance mutations (Figure 3 and Table 2), which are directly responsible for drug resistance, and secondary (compensatory) mutations, which promote or enhance replication competence.53 Compensatory mutations emerge because the selection of resistance-associated changes in the viral polymerase is usually associated with some cost in replication fitness for the virus; these compensatory mutations are important in the context of antiviral resistance because they "fix" the discriminatory primary drug-resistant mutations into the genetic archive of viral cccDNA, the HBV minichromosome, thus providing "quasispecies memory."49 The common mutations that confer resistance to LMV and LdT (eg, rtM204V/I ± rtL180M) confer cross-resistance to other L-nucleosides and reduce sensitivity to ETV but not to ADV or TDF. Conversely, mutants that are resistant to ADV (eg, rtN236T) and TDF generally remain sensitive to L-nucleosides and ETV. Both the L-nucleosides (LMV and LdT) and alkyl phosphonates (ADV and TDF) also select for the mutation rtA181T/V, thereby making it a marker for multidrug resistance. Multiple mutations (eg, rtA184A/A/I/L, rtS202G/L, rtM250I/V) in addition to those that confer resistance to LMV and LdT (rtM204V/I ± rtL180M) are required for high-level resistance to ETV (see Table 2). Cross-resistance across NA groups (eg, rtA181T) (see Figure 3 and Table 2) might eventually be overcome by development of drugs that block stages of the viral life cycle distinct from those inhibited by NA (Supplementary Figure 1). However, such drugs are unlikely to become available for clinical use in the near future. Thus, it is important to understand more fully the molecular mechanisms of NA resistance because, to optimize their use, we must develop methods for defining, detecting, and quantifying drug resistance and cross-resistance.
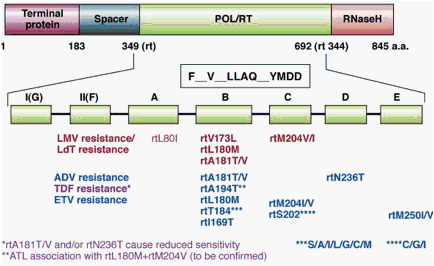
Figure 3. Primary antiviral drug resistance mutations. Polymerase gene mutations conferring resistance to nucleos(t)ide analogs are depicted. Resistance to lamivudine (LMV) and telbivudine (LdT) is conferred by mutations in the YMDD motif within the C domain of the polymerase, ie, rtM204V or rtM204I, often associated with compensatory mutations in the B domain restoring a higher replication capacity, ie, rtL180M and/or rtV173L. Resistance to adefovir (ADV) is conferred by a rtA181V or rtA181T substitution or a rtN236T substitution. The rtA181V/T substitution can also confer decreased susceptibility to LMV and LdT. Resistance to entecavir (ETV) is conferred by a combination of mutations in the B, C, or D domain of the viral polymerase, in addition to a background of substitutions at position rt204. Resistance to tenofovir (TDF) may be conferred by amino acid substitution at position rt194, which needs to be confirmed.
Factors That Predispose to Resistance
There are several major risk factors for development of resistance to NA, especially to LMV. These include a high level of HBV DNA, high serum levels of ALT, and high body mass index.3, 5, 54 Prior therapy with NA, as well as inadequate viral suppression during therapy, has also been shown to predict drug resistance.3, 4, 9, 11 Transmission of drug-resistant mutants in newly infected patients is also likely to predispose to more rapid resistance once treatment is initiated, as it was shown for HIV infection.
Pathways of Resistance
The molecular mechanisms of resistance to drugs for CHB have been recently reviewed in this Journal,55 and resistance tends to be NA structure (sugar residue) specific, providing a structural framework for resistance selection.
L-Nucleoside-Associated Resistance
Resistance to LMV and LdT has been mapped to the YMDD locus in the catalytic (C domain) of HBV Pol,56 mediated primarily by the mutations rtM204I/V (domain C) ± rtL180M (domain B) and rtA181T/V57 (Figure 3). Compensatory mutations that increase viral replication levels can be found in other domains of the HBV Pol, such as rtL80V/I,58 rtI169T,59 rtV173L,60 rtT184S/G, rtS202I, and rtQ215S.61
The mutations rtM204V/I do not confer cross-resistance to ADV or TDF (Table 2), whereas rtA181T/V does.61, 62 The mutations rtI169T, rtT184S/G, and rtS202I/G contribute to ETV resistance but do not confer significant resistance on their own59, 63; rtM204V/I and rtA181T are cross resistant with all other L-nucleoside analogues tested including LdT (USA product insert; FDA, Washington, DC) (Table 2 and Figure 3). Mutations that confer LMV resistance decrease in vitro sensitivity of hepatocytes to the drug by 100- to more than 1000-fold. rtM204I has been detected in isolation, but rtM204V and rtM204S are found only in association with other changes in the A or B domains.64 The pattern of pol sequence in which resistance mutations are usually detected include the following: (1) rtM204I, (2) rtL180M+rtM204V, (3) rtL180M+rtM204I, (4) rtV173L+rtL180M+rtM204V, and (5) rtL80V/I ± rtL180M+rtM204I; the dominance of a particular mutation sequence is associated with HBV genotype.3, 65
Alkyl Phosphonate-Associated Resistance
Resistance to ADV was initially associated with mutations in the B (rtA181T/V) and D (rtN236T) domains of the enzyme62, 66, 67 (Figure 3). These substitutions result in only a modest (3- to 8-fold) increase in the concentration of the drug required for 50% inhibition for viral replication in vitro (effective concentration [EC50]) and are partially cross resistant with TDF (Table 2) almost certainly because of their similar chemical structures.61 The mutation rtN236T does not significantly affect sensitivity to LMV, LdT, or ETV66, 67 but decreases the efficacy of TDF in vitro.68 rtA181T/V confer decreased susceptibility to ADV and TDF and are partially cross resistant to LMV62 and LdT (Table 2). Another mutation (rtI233V) has been recently identified that appeared to confer resistance to ADV.69 In clinical studies, the rtI233V mutation occurred in approximately 2% of all patients with CHB,69 but the final significance of this mutation does require independent confirmation because other groups have not found an association between rtI233V and ADV resistance.70, 71, 72
Tenofovir was originally approved for the treatment of human immunodeficiency virus (HIV)-acquired immunodeficiency syndrome (AIDS) and has also been used to treat patients with HIV-HBV coinfection. Genotypic resistance to TDF has been detected in several patients with HIV-HBV coinfection; the substitution rtA194T (plus rtL180M+rtM204V) has been associated with TDF resistance73; however, a recent report failed to confirm this74; therefore, further studies are needed. The detection of rtA181T/V and rtN236T in patients failing ADV therapy resulted in reduced antiviral efficacy when patients were switched to TDF. Van Bommel et al75 demonstrated an intensification and consolidation of these ADV-resistant clones following this switch and also reduced antiviral efficacy of TDF.
D-Cyclopentane-Associated Resistance
Mutations in HBV polymerase associated with the emergence of ETV resistance have been mapped to the B domain (rtI169T, rtL180M, and/or rtS184G), C domain (rtS202I and rtM204V), and E domain (rtM250V) of HBV Pol (Figure 3). In the absence of the LMV-resistance mutations rtL180M and rtM204V/I, the mutation rtM250V increases the median EC50 of ETV by 10-fold, whereas the rtI169T, rtT184G, or rtS202I have only a modest effect on IC50 values.59, 61, 76, 77, 78, 79 Three other mutations in HBV Pol (rtL180M+rtM204V and either rtT184G/S or rtS202I/G or rtM250V) are required for ETV resistance to develop (Figure 3).
Mutational Pathways and Cross-resistance
Eight codons in HBV polymerase are thus associated with primary drug resistance to NA: 169, 180, 181, 184, 202, 204, 236, 250. These 8 codons have been shown to be involved in HBV antiviral drug resistance via 4 pathways of viral evolution80 (Table 2). (1) the rtM204V/I pathway for L-nucleosides; (2) the rtN236T pathway for alkyl phosphonates; (3) the rtA181T/V pathway, which is shared between the L-nucleosides and alkyl phosphonates; and (4) the D-cyclopentante/entecavir pathway (rtL180M+rtM204V+I169T+T184S/G/C+S202C/G/I+M250I/V).
The first 3 pathways are associated with only 1 mutation, whereas the fourth pathway requires at least 3 mutations for resistance. This "pathways of evolution approach" facilitates understanding HBV evolution during NA therapy and can be used to predict patient outcomes and improve our understanding of cross-resistance patterns and profiles.80
Multidrug Resistance
Sequential monotherapy can promote selection for multidrug-resistant (MDR) strains of HBV, especially when patients are sequentially treated with drugs with similar characteristics, such as with LMV followed by ETV63, 81 or LMV followed by ADV.37, 68 Clonal analyses have shown that MDR usually occurs via the sequential addition of resistance mutations to the same viral genome; mutants that arise from this selection process have full resistance to both drugs. Studies have shown that MDR strains arise if an "add-on" therapeutic strategy does not result in rapid and complete viral suppression, especially if there is a large replication space available for the mutants to spread (ie, necroinflammatory activity or high levels of serum ALT, resulting in hepatocyte proliferation or need for a liver graft). A longitudinal clonal and phenotypic analysis of variants in a patient with a MDR strain of HBV after liver transplantation revealed mutations in the overlapping polymerase and surface genes that conferred resistance to both LMV and ADV as well as a decreased recognition of the virus by anti-HBs antibodies.37 These findings emphasize the need to achieve complete viral suppression during antiviral therapy.
Some specific single mutations confer MDR. This was shown with the rtA181V/T substitution, which is responsible not only for decreased susceptibility to the L-nucleosides LMV and LdT but also to the alkyl phosphonates ADV and TDF.43, 62 This emphasizes the need for genotypic testing in patients with treatment failure to determine the resistance mutation profile and tailor therapy to the major viral strain circulating in the patient. Studies of the antiretroviral agents used to treat HIV have shown that drug resistance testing can be used to monitor response to therapy and aide in the selection of new drug regimens for patients who have failed to respond to antiviral therapy.82
Detection and Monitoring of Resistance
Viral Load Assays
Measurement of viral load is indispensable for monitoring and confirming the presence of drug-resistant virus because nearly all instances of resistance to NA are initially identified by a sustained rise in viral load that occurs despite continuing antiviral therapy. The sensitive HBV DNA assays that are currently in use will detect rising viral loads because of drug-resistant virus even when the emergence of the drug-resistant HBV population is slow. Because factors other than drug resistance (for example, poor patient compliance and/or pharmacogenomic factors) can affect viral load, it cannot be automatically assumed that rising loads are indicative of drug resistance because drug-resistant HBV can only be confirmed by genotyping and/or phenotyping.
Genotyping
To identify potential genotypic resistance, the nucleotide and deduced amino acid sequence of the HBV polymerase isolated from the patient during virologic breakthrough should be compared with the sequence of HBV isolated from a pretherapy sample from the same patient.83 When pretherapy samples are not available for analysis, sequence data at the time of virologic breakthrough should be compared with consensus published sequences(s) of the same HBV genotype.72
Genotyping relies on either DNA sequencing or hybridization. Sequencing-based methods include standard population-based polymerase chain reaction (PCR), cloning of PCR products, and restriction fragment-length polymorphism analyses. Direct PCR-based DNA sequencing can detect a particular mutant only if it is present ≥20% of the total quasi species pool.61 Cloning can overcome this problem, but analysis of large numbers of clones is required. Viral mutants that constitute as little as 5% of the total population can be detected by restriction fragment-length polymorphism analyses, but separate sets of endonuclease reactions must be designed specifically for each (and known) mutant of interest. These methods are labor intensive, require highly skilled personnel, and are not suitable for high-throughput screening. They are used only for "in house" or "home-brew" assays; with the exception of the TRUGENE genotyping test developed by Visible Genetics (Siemens Healthcare Diagnostics, Tarrytown, NY), few have been commercialized or approved by regulatory bodies.
Pyrosequencing
Pyrosequencing is a new sequencing method that relies on the detection of DNA polymerase activity by measuring the pyrophosphate (PPi) released by the addition of a dNMP to the 3' end of a primer. It allows determination of the sequence of a single DNA strand by synthesizing a complementary strand, 1 base pair at a time, and detecting which base was added at each step. Currently, the main limitation of pyrosequencing is that the maximum length of individual sequencing runs are shorter than those obtainable with conventional chain termination sequencing methods. Pyrosequencing is currently the fastest and probably most sensitive (0.1%) method available for detecting small subpopulations of resistant virus84, 85 and is likely to become the method of choice in the near future, particularly if the associated instrumentation becomes more affordable.
Hybridization
Examples of hybridization-based genotyping methods, which can detect single nucleotide mismatches include the following:
1. Mass spectrometric (matrix-assisted laser-desorption ionization time of flight mass spectrometry [MALDI-TOF MS]) analysis of small DNA fragments that can identify mutants that constitute as little as 1% of the total viral population.86
2. The commercially available line probe assay, (INNO-LiPA, Innogenetics, Ghent, Belgium) which relies on the differential hybridization of particular targets to a series of short membrane-bound oligonucleotide probes to discriminate between wild-type sequences and those of known drug-resistant mutants.87 LiPA assays can detect developing viral resistance when the mutants responsible constitute only a minor fraction of the total viral population (5%-10%), an advantage in cases in which there is a high risk of disease progression.88
3. DNA chip technologies. Sequencing with microchip-based technology using oligonucleotide microarrays has the clear advantage of improved sensitivity as well as ability to detect "new" mutants.89 These assays are relatively easy to perform for the simultaneous detection of a multitude of unique mutations as well as recognized polymorphisms.90
One of the main limitations of all hybridization-based methods is their specificity: new sets of specific probes are required for every mutant, and natural sequence variability in regions of interest reduces their discriminatory power and specificity. Furthermore, sequence context and secondary structures in the target can affect sensitivity, and minor subpopulations (those constituting less than 10% of the total population) may escape detection. For detection of known and "new" mutants, genotyping using oligonucleotide microarrays appears to be the only viable alternative to direct sequencing, but, because the number of clinically relevant HBV mutants is still relatively small and the technology is specialized and expensive, it will be some time before they become cost-effective.
In Vitro Phenotypic Assays
Several approaches have been developed to perform in vitro phenotypic analysis of the resistant mutants identified in vivo in patients. These assays are critical to determine the role of a given mutation profile in drug resistance as well as to determine the cross-resistance profile of those mutants. These approaches include viral polymerase enzymatic assays, cell lines permanently expressing HBV resistant mutants, and cell culture models in which the viral genome of resistant mutants is transferred for the analysis of viral replication and drug susceptibility; these assays have been reviewed recently.91 All these assays have been useful to demonstrate the role of a given polymerase gene mutation, observed in patients with treatment failure, in the development of antiviral drug resistance. They are also important to determine the cross-resistance profile of the main resistance mutations (see Table 2)51, 92 and help develop clinical management algorithms.53, 93, 94 Further useful information is also generated on viral fitness, which is an important determinant in the process of understanding the patterns and profiles of resistant mutant selection in the patient.
Clinical Aspects of Resistance
Definitions
All patients receiving NA therapy for CHB should be closely monitored for virologic response and breakthrough during treatment and for durability of response and viral relapse after treatment has stopped.53 Serum HBV DNA should be tested every 3 months during treatment.93 Failure of antiviral therapy of CHB may follow different directions, which rely on specific mechanisms and therefore have clinical implications in terms of treatment adaptation. Thus, it is important to distinguish between primary nonresponse, partial virologic response, and virologic breakthrough because of antiviral drug resistance (Figure 1).
Primary nonresponse
The failure to achieve a 1-log10 copies/mL (or 1.0 log10 IU/mL) decline in viral load after 12 weeks of therapy is considered as a primary nonresponse.53, 93, 94 It may be due to a problem of compliance or the medication may not exhibit its antiviral activity in a given patient. Suboptimal response has been shown to be due to host pharmacologic effect and/or to patient compliance but not to a reduced susceptibility of viral strains to ADV as measured in vitro by phenotypic assay.70 With more potent antiviral drugs, this phenomenon seems to be less frequent. When a primary nonresponse is identified, antiviral treatment should be modified to prevent disease progression and subsequent risk of emergence of populations of drug-resistant mutants. The week 12 time point of therapy is therefore important to determine the antiviral activity of the treatment regimen and assess treatment adherence.
Virologic breakthrough: viral rebound
Virologic breakthrough typically results from the emergence of drug-resistant viral strains. It is defined by an increase of at least 1-log10 copies/mL (or >1.0-log10 IU/mL) compared with the lowest value (or nadir) during treatment, confirmed by a second test, in a treatment compliant patient.53, 93, 94 Depending on the mutation profile selected by the drug, viral load increase may be slow, making the diagnosis of rebound difficult. It usually follows the detection of genotypic resistance (Figure 1), ie, detection of resistance mutations.3, 53, 93 In the absence of treatment adaptation, the rise in viremia levels may be followed in subsequent weeks or months by an increase in ALT levels (biochemical breakthrough) and subsequently progression of liver disease (clinical breakthrough).
Treatment of HBV Drug Resistance
Primary nonresponse
A primary nonresponse is observed more frequently in patients treated with ADV (approximately 10%-20% of patients) than in those treated with other NA, probably because patients are inadequately dosed.70 Patients who do not respond to ADV should be rapidly switched to TDF or ETV therapy. A primary nonresponse to LMV, LdT, ETV, or TDF is observed only rarely2; in these patients, it is important to determine the level of compliance. If a patient with a primary nonresponse to these drugs is compliant, analysis of HBV NA-resistance mutations can identify alternate treatment strategies96 (see Table 2).
Partial virologic response
Partial virologic responses have been observed with all NA used in CHB. Again, it is important to check for compliance. There are 2 strategies for treating patients who have a partial virologic response to LMV, ADV, or LdT at week 24: change to a more potent drug (ETV or TDF) or add a more potent drug that does not share cross-resistance. Tenofovir should not be added to ADV therapy if the patient is infected with an HBV mutant that is resistant to ADV (ie, rtA181T/V ± rtN236T) because these drugs belong to the same chemical group of NA, the alkyl phosphonates.15, 96, 99, 100
Virologic breakthrough
Virologic breakthrough in compliant patients is related to viral resistance. Resistance is associated with prior treatment with NA or, in treatment-naïve patients, with high baseline levels of HBV DNA, a slow decline in HBV DNA levels, and partial virologic response to treatment. Resistance should be identified as early as possible, before ALT levels increase, by monitoring HBV DNA levels and if possible identifying the NA resistance profile; the therapeutic strategy can be determined based on this information. Clinical and virology studies have demonstrated the benefit of an early (as soon as viral load increases) adaptation of treatment.95, 96, 101 In cases of resistance, an appropriate rescue therapy should be initiated that has the most effective antiviral effect and minimal risk for selection of MDR strains. Therefore, adding a second drug that is not in the same cross-resistance group as the first is the recommended strategy.
Table 2 shows the cross-resistance data for the most frequent resistant HBV variants.92, 96 Treatment adaptation should be performed accordingly and is summarized as follows:
· LMV resistance: add TDF (add ADV if TDF not available);
· ADV resistance: it is recommended to switch to TDF if available AND add a second drug without cross-resistance. If an rtN236T substitution is present, add LMV, ETV, or LdT or switch to TDF plus emtricitabine. If an rtA181V/T substitution is present, it is recommended to add-on ETV or to switch to TDF plus ETV or TDF plus emtricitabine (as a single tablet: Truvada);
· LdT resistance: it is recommended to add TDF (or ADV if TDF is not available);
· ETV resistance: it is recommended to add TDF;
· TDF resistance: primary resistance to TDF has not been confirmed so far. It is recommended that genotyping and phenotyping be done by a reference-type laboratory to determine the cross-resistance profile. Entecavir, LdT, LMV or emtricitabine could be added but would depend on the profile (refer to Table 2).
Note that the safety of some combinations in the longer term is presently unknown and that add-on therapy is not always successful in achieving adequate viral inhibition (PCR undetectability).
Preventing Resistance
The spread of drug-resistant HBV mutants can be reduced by avoiding unnecessary drug use, choosing drugs and combinations more carefully, and continually monitoring or carrying out targeted surveillance for drug resistance.45
Because of the unusual replication strategy used by HBV, viral populations are genetically heterogeneous, so even treatment-naïve patients have drug-resistant mutants that constitute only a minor component of the population in the absence of selection pressure from antiviral drugs. A majority of patients may not require antiviral therapy. Several professional bodies (including the American Association for the Study of the Liver, the European Association for the Study of the Liver, the Asian Pacific Association for the Study of the Liver, and the National Institutes of Health) publish regularly updated guidelines to assist clinicians with recognition, diagnosis, prevention, and management of CHB: these are unanimous in recommending that therapy should be considered for patients with only more active or advanced liver disease and others most likely to respond in the context of defined treatment end points. Treatment algorithms have been developed to assist in identification of suitable candidates for treatment and to determine when to initiate treatment.
Because drug-resistant mutant HBV populations are established and expand through replication, antiviral therapy, once initiated, should aim to suppress viral replication as completely and rapidly as possible. The lower risk of resistance to TDF and ETV (compared with LMV, LdT, and ADV) supports their use as first-line therapy, especially in patients who have received liver transplants and those with cirrhosis or decompensated liver disease because development of drug resistance is more likely to precipitate clinical deterioration in these groups.
Combination chemotherapy is being used more frequently to treat CHB. It is effective when the appropriate combinations are employed and can reduce the risk of drug resistance. Although HBV mutants that are resistant to single drugs exist before therapy starts and can evolve rapidly in patients, HBV mutants with MDR are much less likely to exist before treatment. Ideally, drugs used in combination should have different mechanisms so that they have additive/synergistic effects. Combination therapy using NA with a complementary cross-resistance profile prevents the development of resistance but does not have increased antiviral effects, compared with single-drug therapy.102 Use of interferon in combination with NA is probably the next logical step. Although initial clinical trials of such combinations were disappointing, results from later trials are more encouraging. However, the added benefit of the combination tends to be lost after treatment cessation.103, 104
Combinations of L-nucleosides are unlikely to be more effective than therapy with single L-nucleosides and can have antagonistic effects (because they compete for cellular activation mechanisms and viral targets). The lack of cross-resistance of HBV mutants to LMV and ADV observed in vitro (except for rtA181T/V) and in some clinical studies indicates that these drugs could be effective in combination. Preliminary data also support the use of ETV in combination with ADV or TDF, but definitive recommendations will require further clinical trials and cost-benefit studies.
Each patient's response to treatment should be monitored carefully so that drug resistance can be detected early, before viral breakthrough and disease progression. Assays for serum levels of HBV DNA and ALT should be performed 3-6 months after therapy begins, to check for efficacy and compliance; lack of compliance is the most common cause of primary treatment failure. Additional assays, performed at 6-month intervals during the first 2 years of treatment, are recommended for patients with mild liver disease. Patients should then be assessed for viral load and ALT level every 3 months after 2 years of therapy: this is the time during which the probability of developing resistance increases. The consequences of resistance appear more rapidly and can become life-threatening in patients with advanced disease; these patients should be tested for viral load and ALT level every 3 months. Once the viral load increases to ≥1.0-log10 IU/mL, HBV Pol should be sequenced to identify resistance mutations and determine the next therapeutic approach, based on cross-resistance information (Figure 4).
Figure 4. Management flow chart for first virologic breakthrough/partial virologic response.

Infectivity and Public Health Aspects
The gene that encodes the HBV polymerase overlaps with the gene that encodes the viral envelope, and so mutations in the overlapping reading frame can change both proteins (Figure 5A). The nucleotide change that alters codon rt204 (rtM204I/V) in the polymerase gene confers resistance to LMV, LdT, and ETV and also results in a nonsynonymous change in the gene encoding the hepatitis B surface antigen (HBsAg), directly in the overlapping region. The rtM204V mutation typically results in the substitution sI195M in HBsAg, whereas the rtM204I change can cause sW196S, sW196L, or a termination codon (Figure 5B), depending on codon useage. The mutation rtL180M is synonymous in HBsAg, but rtV173L results in sE164D, and this combination of mutations is found in up to 20% of cases of LMV resistance.3, 65 The ADV resistance mutation rtN236T overlaps with the stop codon at the end of the envelope gene and does not affect HBsAg. The mutation selected by ADV and/or LMV/LdT at rtA181T typically results in a stop mutation in the envelope gene (sW172stop) (Figure 5B), and the ADV resistance mutation at rtA181V results in a concomitant change sL173F. Mutations that result in a stop codon mutation in the envelope gene, such as those for LMV, LdT (rtM204I and rtA181T), and ADV (rtA181T), are usually found in the presence of a low percentage of wild-type HBV to ensure rescue of the mutant by the wild-type to allow viral packaging and release of the defective variant.43 The ETV resistance mutation rtI169T results in a change at sF161L. This mutation, along with sE164D, is located within the region that is defined as the "a" determinant, which includes amino acids 95 to 165-the major antibody neutralization domain of HBV.
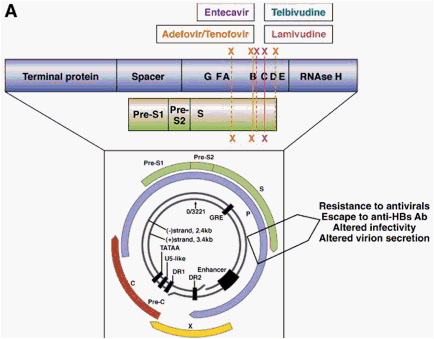
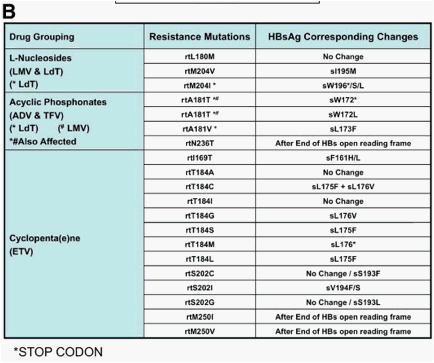
Figure 5. Impact of drug resistance mutations in the viral polymerase gene on the overlapping surface gene. Panel A: Physical map showing the impact of drug resistance mutations in the viral polymerase gene on the envelope gene. Resistance mutations may therefore result in viral envelope changes leading to altered virion secretion, altered infectivity, and escape to anti-HBs antibodies. Panel B: Antiviral drug-associated HBsAg changes. The main amino acid substitutions in the viral polymerase and their corresponding changes in the envelope proteins are shown.
Thibault et al published the first case report of primary infection with LMV-resistant HBV105 (rtL180M+rtM204V), which was associated with a typical bout of acute hepatitis and an incubation period of 2-3 months. The level of viremia was lower than that usually observed during the acute phase of hepatitis B, and the virus was cleared. The acutely infected individual had not been previously vaccinated against HBV.
Several studies have reported mutations in HBsAg that alter its antigenicity. Torresi et al observed that the LMV resistance mutations rtV173L+rtL180M+rtM204V (producing sE164D and sI195M in HBsAg) resulted in reduced binding of antibody to this antigen,106 although the reduction was not as great as that caused by the mutant sG145R, compared with wild-type HBV. These results were confirmed and extended by Sloan et al using cell-derived HBVs.107
Very few in vitro studies have been performed to study the infectivity of the NA-resistant mutants. The combination of polymerase and surface gene mutations might result in viruses that exhibit a reduced fitness that translates to differences of selection kinetics. However, these studies are hampered by the challenges of working with primary human hepatocytes, the only cellular system available for these investigations. Bartholomew et al infected primary human hepatocytes with HBV using serum samples collected from patients before the start of treatment with LMV and after viral breakthrough.108 Their results demonstrated that the viral strains isolated at the time of viral breakthrough were resistant to LMV but could still infect hepatocytes. The hepatocyte progenitor cell line HepaRG has specific hepatocyte functions and can be infected by HBV; primary cultures of normal human hepatocytes were also used in infectivity studies of clinical isolates, especially of drug-resistant strains.109 These studies showed that mutations in the HBV polymerase and overlapping surface genes can impair replication capacity, virion secretion efficiency, and infectivity;42 some of these mutant HBVs escaped antibody recognition and could therefore mediate breakthrough infection in vaccinated individuals and escape detection by commercial diagnostic kits. The effect of these substitutions on HBV infectivity was shown to be a critical determinant of which resistant mutants would spread more rapidly in the liver and dominate other HBV variants.42
In chimpanzees, Kamili et al44 challenged the immunity induced by a commercial hepatitis B vaccine against a tissue culture-derived HBV clone that contained 3 polymerase mutations (rtV173L, rtL180M, rtM204V) and substitution mutations in the overlapping region that encodes the envelope/HBsAg (sE164D, sI195M). Immunologic evidence of HBV infection and replication was observed in the vaccinated chimpanzees after challenge with the mutant as well as after rechallenge with serum-derived wild-type HBV, despite robust humoral and cellular anti-HBV immune responses to the vaccine. The observed infection by the mutant form of HBV, despite the presence of high levels of HBV antibody (which were considered to be protective), are consistent with clinical reports of breakthrough infections in anti-HBs-positive patients who are infected with escape HBV mutants.44 Therefore, "antiviral drug-associated potential vaccine escape mutants" have the potential to jeopardize the hepatitis B immunization program110 (Supplementary Figure 2). As more HBV mutants arise that have resistance to different antiviral agents, the effects on the antigenicity of the HBsAg protein will need to be established.
Future Directions and Conclusion
Very few new drugs are being developed to treat HBV; therefore, it is important to continue research into mechanisms of pathogenesis and resistance and to identify new therapeutic targets. Small molecule inhibitors that are directed against multiple HBV targets should improve viral clearance and prevent resistance (see Supplementary Figure 1). For example, virus entry into the cell can be inhibited111, 112 using pre-S1 peptides, which mimic the envelope protein domain involved in virus-cell membrane interaction. These peptides prevented HBV entry into cultured hepatocytes and inhibited subsequent viral infection and spread in the HBV animal model of severe combined immunodeficient mice.112 Combination of these peptides with NA could prevent the infection of new cells when viral load is suppressed by NA and increase the rate of clearance of infected cells as well as preventing further de novo infections. Ongoing preclinical and clinical studies will determine their efficacy and safety.
Reagents have been tested that target cccDNA or steps in its formation and processing, but these have been found to have cytotoxic effects. Agents that modify epigenetic regulation of cccDNA transcriptional activity are being investigated in experimental models.113 Viral pregenome encapsidation and capsid formation also represent potential targets. Phenylpropenamide derivatives and heteroaryl-pyrimidines are known to inhibit the replication of wild-type and LMV-resistant mutant genomes in hepatoma cell lines,114, 115, 116, 117 although clinical trials were not conducted because of formulation problems. AiCuris Pharmaceuticals (Wuppertal, Germany) has developed the heteroaryl-pyrimidines molecules further as non-nucleoside inhibitors of HBV core protein dimerization that prevent nucleocapsid formation.116, 117 The heteroaryl-pyrimidines prevented HBV infection in an animal model116 and represent a potentially important new advance in chemotherapy.
Viral morphogenesis and egress are also useful targets. Iminosugars, which modulate the glycosylation status and conformation of envelope proteins, can decrease the production of infectious particles in vitro.118 They were shown to have an antiviral effect in the woodchuck model of hepadnavirus infection.119 Therapeutics might also be developed to modulate the innate response of infected hepatocytes,120, 121 dendritic cell activation,122 or the adaptive immune response.123, 124, 125 Induction of sustained immunologic control of HBV infection could allow for timed cessation of NA administration.
Ideally, treatment for CHB should begin at diagnosis; this is not feasible because of limitations of drugs. Clinical trials and concurrent improvements in diagnostic technology ensure that treatment options and expert opinion on patient management will continue to evolve. Many laboratories are genotyping and phenotyping HBV mutants to delineate patterns of resistance and cross-resistance. These data will improve the design of new therapeutic strategies and maximize the benefits of antiviral agents. Resistance-testing methodologies vary, and, although few direct comparisons have been made, in vitro phenotype testing (if possible, in combination with genotype testing) seems superior to conventional genotype or virtual phenotyping testing. This is especially true for analysis of HBV isolated from patients who have already been treated with several drugs, in whom multiple mutations are more likely to have become fixed in the genetic archive. Drug resistance testing of HBV isolates is currently performed only in a few specialized "reference-type" laboratories, but it should become routine as more sensitive, reliable, high-throughput, and accurate methods are developed, along with clinically useful algorithms for interpretation. In particular, standardization of tests and definition of resistance/susceptibility thresholds or "cut-offs" that can be used to correlate laboratory results with clinical observations and outcomes are urgently needed.
Methods for assessing the relative replication capacity of HBV mutants are being developed and will prove useful in selecting therapy and tailoring individual patient management. In cases in which treatment failure cannot be attributed to patient noncompliance or emergence of resistant virus, host factors might be involved. It is important to monitor viral load throughout therapy so that the treatment strategy can be modified in cases of partial response or virologic breakthrough.
The choice of the first-line agent(s) is very important; the goal is to delay the development of resistance and to preserve treatment options over the long-term of treatment for CHB. The development of therapeutics that complement the effects of NA is warranted; new treatment strategies must be aimed at increasing viral suppression and promoting virologic clearance and ensuring the prevention of drug resistance and its complications.
|
|
| |
| |
|
|
|