| |
Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial -- attached is pdf and appendix with adverse events
|
| |
| |
Download PDF: Number (%) of Patients with Specific Serious Clinical Adverse Experiences of Any Cause by Organ System Category*
Download PDF: Safety and effi cacy of raltegravir-based versus
efavirenz-based combination therapy in treatment-naive
patients with HIV-1 infection: a multicentre, double-blind
randomised controlled trial
The Lancet, Early Online Publication, 3 August 2009
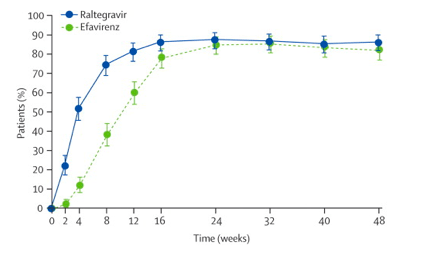
indicating that raltegravir was non-inferior to efavirenz (p<0·0001 for non-inferiority.....The main analysis (with non-completion counted as failure) showed that 86·1% (n=241 patients) of the raltegravir group and 81·9% (n=230) of the efavirenz group achieved the primary endpoint (difference 4·2%, 95% CI -1·9 to 10·3).....The primary analysis was per protocol. The margin of non-inferiority was 12%. We also analysed the data using the observed-failure method, which confirmed that raltegravir was non-inferior to efavirenz for achievement of the primary endpoint (p<0·0001 for non-inferiority; table 2). More patients achieved viral suppression to less than 50 vRNA copies per mL in the raltegravir group than in the efavirenz group at early time points (weeks 2-16). The time to achieve such viral suppression was shorter for patients on raltegravir than those on efavirenz (log-rank test p<0·0001). The mean change in CD4 cell count from baseline to week 48 was 189 cells per µL (95% CI 174-204) for raltegravir recipients and 163 cells per µL (148-178) for efavirenz recipients (difference 26 cells per µL, 95% CI 4-47, p=0·0184.
Prof Jeffrey L Lennox MD a, Edwin DeJesus MD b, Prof Adriano Lazzarin MD c, Prof Richard B Pollard MD d, Jose Valdez Ramalho Madruga MD e, Daniel S Berger MD f, Jing Zhao PhD g, Xia Xu PhD g, Angela Williams-Diaz BS g, Anthony J Rodgers MS g, Richard JO Barnard PhD g, Michael D Miller PhD g, Mark J DiNubile MD g, Bach-Yen Nguyen MD g, Randi Leavitt MD g, Dr Peter Sklar MD g Corresponding AuthorEmail Address, for the STARTMRK investigators
Summary
Background
Use of raltegravir with optimum background therapy is effective and well tolerated in treatment-experienced patients with multidrug-resistant HIV-1 infection. We compared the safety and efficacy of raltegravir with efavirenz as part of combination antiretroviral therapy for treatment-naive patients.
Methods
Patients from 67 study centres on five continents were enrolled between Sept 14, 2006, and June 5, 2008. Eligible patients were infected with HIV-1, had viral RNA (vRNA) concentration of more than 5000 copies per mL, and no baseline resistance to efavirenz, tenofovir, or emtricitabine. Patients were randomly allocated by interactive voice response system in a 1:1 ratio (double-blind) to receive 400 mg oral raltegravir twice daily or 600 mg oral efavirenz once daily, in combination with tenofovir and emtricitabine. The primary efficacy endpoint was achievement of a vRNA concentration of less than 50 copies per mL at week 48. The primary analysis was per protocol. The margin of non-inferiority was 12%. This study is registered with ClinicalTrials.gov, number NCT00369941.
Doses of raltegravir were taken 10-14 h apart without regard to food intake, whereas efavirenz was taken on an empty stomach at the hour of sleep. To ensure masking of treatment, patients in the raltegravir group were given placebo tablets identical to efavirenz and vice versa. All participants also took 300 mg tenofovir and 200 mg emtricitabine as a single tablet in the morning with food.
Findings
566 patients were enrolled and randomly allocated to treatment, of whom 281 received raltegravir, 282 received efavirenz, and three were never treated.
At baseline, 297 (53%) patients had more than 100 000 vRNA copies per mL and 267 (47%) had CD4 counts of 200 cells per µL or less.
The main analysis (with non-completion counted as failure) showed that 86·1% (n=241 patients) of the raltegravir group and 81·9% (n=230) of the efavirenz group achieved the primary endpoint (difference 4·2%, 95% CI -1·9 to 10·3).
The time to achieve such viral suppression was shorter for patients on raltegravir than on efavirenz (log-rank test p<0·0001).
Significantly fewer drug-related clinical adverse events occurred in patients on raltegravir (n=124 [44·1%]) than those on efavirenz (n=217 [77·0%]; difference -32·8%, 95% CI -40·2 to -25·0, p<0·0001).
Serious drug-related clinical adverse events occurred in less than 2% of patients in each drug group.
Interpretation
Raltegravir-based combination treatment had rapid and potent antiretroviral activity, which was non-inferior to that of efavirenz at week 48. Raltegravir is a well tolerated alternative to efavirenz as part of a combination regimen against HIV-1 in treatment-naive patients.
Funding
Merck.
Introduction
Highly active antiretroviral therapy is the standard of care for patients with HIV infection.1, 2 Combination regimens that include inhibitors of viral reverse transcriptase and protease enzymes have proven to be cost-effective interventions resulting in improved survival and decreased morbidity for patients with low CD4 cell counts.3 Although several recommended regimens are available for the initial treatment of HIV-1 infection, the success of antiretroviral therapy continues to be limited by preferences of patients and care-providers, underlying comorbidities, drug interactions, adverse drug effects, and long-term complications. Transmission of resistant HIV-1-particularly strains resistant to non-nucleoside reverse transcriptase inhibitors-places treatment-naive patients at risk of suboptimum responses to standard first-line regimens.4-13 Antiretrovirals from new drug classes are needed to expand options for initial treatment of HIV-1 infection and enhance the probability of achieving and maintaining optimum virological suppression.
Guidelines issued by the US Department of Health and Human Services recommend tenofovir and emtricitabine in combination with either efavirenz or a ritonavir-boosted protease inhibitor as the initial regimen for previously untreated patients.1 The co-formulation tenofovir, emtricitabine, and efavirenz has become the preferred protease-inhibitor-sparing regimen in developed countries because of ease of dosing and potent and durable antiretroviral activity.1,2,14-17
Raltegravir is a novel HIV-1 integrase inhibitor that prevents proviral DNA-strand transfer. The drug has potent in vitro activity against strains of HIV-1 that are susceptible or resistant to other classes of antiretroviral drugs.18-24 In the BENCHMRK studies,20, 21 use of raltegravir with an optimum background regimen was generally well tolerated and provided superior HIV-1 suppression compared with optimum background treatment alone, despite infection with virus resistant to reverse transcriptase and protease inhibitors. Results of a dose-ranging phase II trial in treatment-naive patients showed similar efficacy with raltegravir and efavirenz when each drug was used as part of a combination antiretroviral regimen with two nucleoside reverse transcriptase inhibitors.22 At 96 weeks, 83% of 160 patients on raltegravir and 84% of 38 patients on efavirenz had viral RNA (vRNA) concentration of less than 50 copies per mL.23
We present 48-week results from the phase III STARTMRK study comparing raltegravir-based and efavirenz-based combination regimens as initial treatment for adults infected with HIV-1.
Discussion
We have shown that raltegravir-based combination treatment had rapid antiretroviral activity in treatment-naive patients, and was non-inferior to efavirenz-based combination treatment during the first 48 weeks of treatment. These results were shown for the main analysis in which all patients who did not complete the study were recorded as failures, and were confirmed by analysis with the observed-failure method, indicating that non-inferiority was not solely due to the higher number of discontinuations in the efavirenz group than in the raltegravir group. Both regimens had consistent efficacy across all baseline prognostic and stratification factors, including patients with high viral loads or low CD4 cell counts.
Consistent with earlier studies, significantly more raltegravir than efavirenz recipients achieved less than 50 vRNA copies per mL for weeks 2-16, although the clinical significance of more rapid vRNA decline has not been established.22,26-30 Greater increases in mean CD4 cell count were recorded in the raltegravir group than in the efavirenz group at week 48. Despite this robust response, immune reconstitution syndromes occurred infrequently in this carefully screened study population that included few patients with a reported history of AIDS-defining illness. Virological failure was rare in both treatment groups. Of the eight patients on raltegravir who had virological failure, more than 400 vRNA copies per mL, and an integrase gene that could be amplified, half had mutations known to confer raltegravir resistance. When resistance to raltegravir developed, the mutational pattern was consistent with that previously reported in treatment-experienced patients on raltegravir.20, 24, 31
Raltegravir had a favourable safety and tolerability profile compared with efavirenz. Raltegravir was associated with significantly fewer overall and drug-related clinical adverse events, and CNS-related adverse events than was efavirenz. Neuropsychiatric symptoms generally arise at initiation of efavirenz treatment and, in most patients, improve or disappear during the first few weeks of treatment, although symptoms persist in some patients for months or years.32-38 By comparison, most CNS-related adverse events recorded in our study were mild and transient.
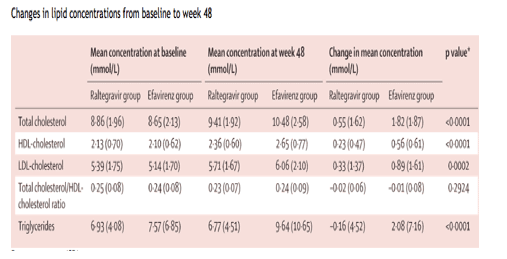
Dyslipidaemias are common complications of antiretroviral drugs, and are often associated with protease inhibitors and reverse transcriptase inhibitors.39-41 Raltegravir had a modest effect on serum lipids between baseline and week 48-small increases were recorded in total cholesterol, LDL-cholesterol, and HDL-cholesterol concentrations, and triglyceride concentration decreased slightly-but changes were significantly higher for efavirenz recipients.
The nucleotide and nucleoside reverse transcriptase inhibitors, tenofovir and emtricitabine, were used in the study because the combination is effective and well tolerated. In a phase I study of healthy individuals and a phase II study of patients with HIV infection, tenofovir slightly increased peak plasma raltegravir concentrations and the area under the raltegravir concentration-time curve.42 On the basis of these results, raltegravir and tenofovir can be given together without dose adjustments. Although raltegravir is expected to have similar effects in combination with reverse transcriptase inhibitors other than tenofovir and emtricitabine (or lamivudine), studies of different raltegravir-based regimens have not yet been done in treatment-naive patients.
Non-nucleoside reverse transcriptase and boosted protease-inhibitor regimens provide effective first-line treatment for most patients with HIV infection, but new options are clearly needed in some circumstances. An efavirenz-based regimen might not be ideal for some patients because of side-effects, teratogenicity, pharmacokinetic interactions, transmitted drug resistance, or a combination of these factors. Resistance to non-nucleoside reverse transcriptase inhibitors in treatment-naive patients varies between countries.4,6-8,11,13,43 In our international study, 2% of patients screened for eligibility were found to have efavirenz-resistant virus, and therefore should not receive efavirenz or nevirapine as first-line treatment.44
Raltegravir is given twice daily, whereas efavirenz-based combination treatment can be given once daily as one pill, which might improve adherence in clinical practice. Our rigorously controlled and blinded study required all participants to take matching placebos in addition to their active drug; such a regimen might have negated the potential advantages of an efavirenz regimen with one pill once per day in terms of convenience and compliance. Other regimens including a protease inhibitor boosted with ritonavir are viable options, but their use could be limited by tolerability, drug interactions, and raised lipid concentrations in some patients. Raltegravir-based regimens have not yet been directly compared with regimens including protease inhibitors as initial treatment for treatment-naive patients.
Our findings that raltegravir-based combination treatment is effective and generally well tolerated in treatment-naive patients, compared with efavirenz, are supported by follow-up data to week 96 in a phase II study of treatment-naive patients.23 Raltegravir is an important additional drug for initial treatment of HIV-1 infection, and should be regarded as an alternative to efavirenz as part of a first-line combination regimen with tenofovir and emtricitabine in treatment-naive patients.
Results
Figure 1 shows the trial profile. 689 patients were screened for the study, of whom 4% were ineligible because of viral resistance to efavirenz (n=17 patients), tenofovir (n=1), emtricitabine (n=3), or tenofovir and emtricitabine (n=1); and for three further patients, results of resistance testing were not available for review. 566 patients were enrolled and 563 (99%) received study drug. 59 (10%) discontinued treatment, mainly because of adverse events. 58% (n=324) of patients were enrolled from non-white ethnic groups and most were men (table 1). More than half (n=297 [53%]) of participants had more than 100 000 vRNA copies per mL. 106 (19%) participants were infected with non-clade B HIV-1. 39 (7%) participants were co-infected with hepatitis B or C. Overall, 267 (47%) patients had a CD4 cell count of 200 cells per µL or less at baseline, of whom 58 (22%) had 50 cells per µL or less (table 1). 98% of participants in both drug groups (276 on raltegravir, 277 on efavirenz) took the study drug on 90% or more of the appropriate days.
In the main analysis that recorded as failures all patients who did not complete the study, 86·1% (n=241) of the raltegravir group achieved the primary endpoint of less than 50 vRNA copies per mL at week 48, compared with 81·9% (n=230) of the efavirenz group (difference 4·2%, 95% CI -1·9 to 10·3), indicating that raltegravir was non-inferior to efavirenz (p<0·0001 for non-inferiority; figure 2). We also analysed the data using the observed-failure method, which confirmed that raltegravir was non-inferior to efavirenz for achievement of the primary endpoint (p<0·0001 for non-inferiority; table 2). More patients achieved viral suppression to less than 50 vRNA copies per mL in the raltegravir group than in the efavirenz group at early time points (weeks 2-16). The time to achieve such viral suppression was shorter for patients on raltegravir than those on efavirenz (log-rank test p<0·0001; figure 3). The mean change in CD4 cell count from baseline to week 48 was 189 cells per µL (95% CI 174-204) for raltegravir recipients and 163 cells per µL (148-178) for efavirenz recipients (difference 26 cells per µL, 95% CI 4-47, p=0·0184; figure 4).
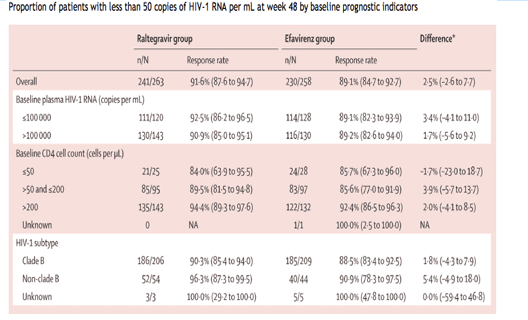
Regimens based on both raltegravir and efavirenz resulted in high achievement of the primary endpoint for patients separated into subgroups of baseline prognostic indicators, including patients with more than 100 000 vRNA copies per mL; numerically more patients with CD4 cell counts exceeding 200 cells per µL responded to treatment with either regimen than did those with lower CD4 cell counts (table 2). Although only a small number of patients were infected with non-clade B HIV-1 subtypes, the results indicated that the proportion of patients who responded to treatment was similar for patients infected with clade B and non-clade B viral subtypes. Increase in CD4 cell count from baseline to week 48, between treatment groups (raltegravir group minus efavirenz group) was greater for patients with a 100 000 vRNA copies per mL or less at baseline (46 cells per µL, 95% CI 19 to 74), than for those with more than 100 000 copies per mL (5 cells per µL, -27 to 37).
Most patients had at least one clinical adverse event during the study, of whom significantly more were on efavirenz than on raltegravir; overall 26 patients discontinued the study as a result, but the proportion of patients who discontinued did not differ significantly between treatment groups (table 3). Significantly more patients on efavirenz than on raltegravir had clinical adverse events that were judged to be drug-related; post-hoc analysis showed that 16% (n=45) and 32% (n=90) of patients on raltegravir and efavirenz, respectively, had drug-related clinical adverse events of moderate-to-severe intensity (p<0·0001). Table 4 shows a full list of drug-related clinical adverse events of moderate-to-severe intensity recorded in more than 2% of patients in either drug group. Specific drug-related clinical adverse events of any severity occurring in more than 10% of participants on either raltegravir or efavirenz were dizziness (6% [n=16] vs 34% [n=95]), headache (9% [n=25] vs 14% [n=39]), and abnormal dreams (7% [n=19] vs 13% [n=37]). Immune reconstitution syndrome was reported as an adverse event in 17 (6%) raltegravir recipients and 11 (4%) efavirenz recipients.
Analysis at week 8 showed that at least one CNS-related adverse event had occurred in 10% (n=29) of patients on raltegravir versus 18% (n=50) of those on efavirenz (p=0·0149). Retrospective sensitivity analysis with additional neuropsychiatric symptoms showed that 20% (n=57) versus 52% (n=147) of raltegravir and efavirenz recipients, respectively, had had at least one CNS-related adverse event by week 8 (p<0·0001). Although most neuropsychiatric symptoms were self-limited, significantly fewer patients on raltegravir than on efavirenz had had one or more CNS-related adverse events by week 48 according to both the main (14% [n=38] vs 23% [n=65], p=0·0044) and sensitivity (26% [n=73] vs 59% [n=165], p<0·0001) analyses of cumulative incidence. In the analysis with the expanded list of clinically relevant neuropsychiatric symptoms from baseline to week 48, all CNS-related adverse events were classed as mild in 46 of 74 (62%) raltegravir recipients and 132 of 167 (79%) efavirenz recipients experiencing neuropsychiatric symptoms. Only one patient, who was on efavirenz, discontinued the trial because of CNS-related adverse events.
Serious adverse events occurred at a similar frequency in both treatment groups (table 3); for a full list of serious clinical adverse events see webappendix pp 1-2. New or recurrent cancers were diagnosed in one (<1%) raltegravir recipient, who had Kaposi's sarcoma, and nine (3%) efavirenz recipients, who had Kaposi's sarcoma (n=6), B-cell lymphoma (n=1), squamous cell carcinoma of the anus (n=1), and bone cancer (n=1). Two patients on raltegravir died during masked treatment; the causes of death were Kaposi's sarcoma and cerebral haemorrhage and were judged to be unrelated to the study drug.
Fewer laboratory-associated adverse events were recorded in patients on raltegravir than on efavirenz, but the difference was not significant (table 4). Specific drug-related laboratory-associated adverse events that occurred in 2% or more patients on either raltegravir or efavirenz were raised concentrations of serum aspartate aminotransferase (6 patients vs 8) and alanine aminotransferase (5 vs 10). Only one patient, who was on efavirenz, had a serious laboratory-associated adverse event of increased concentrations of aspartate aminotransferase and alkaline phosphatase, but this was judged not to be drug-related. Table 5 shows laboratory-associated abnormalities of grade 3 or 4. Overall, adverse events occurred with similar frequency in patients with or without hepatitis B or C co-infection in both treatment groups, except for mild increases in serum aminotransferase concentrations, which were slightly more common in co-infected patients.
At week 48, the mean changes from baseline in total cholesterol, LDL-cholesterol, HDL-cholesterol, and triglyceride concentrations were smaller for raltegravir than for efavirenz recipients (table 6), and the change in the total cholesterol/HDL-cholesterol ratio between treatment groups was not significant. A small decline in triglyceride concentration was noted in raltegravir recipients. In 14 patients who were not being treated at study entry for hyperlipidaemia (n=3 on raltegravir, n=11 on efavirenz), concomitant lipid-lowering treatment was added during the first 48 weeks of the study. Of 21 patients on lipid-lowering treatment at entry, four patients on raltegravir and four on efavirenz received an increased dose or a second drug, or both during the study.
Overall, 66 patients (10% on raltegravir vs 14% on efavirenz) did not achieve less than 50 vRNA copies per mL during the study, defined as virological failure (figure 5). 16 patients with virological failure had more than 400 vRNA copies per mL, allowing genotypic susceptibility tests to be done. One case in which vRNA could not be amplified was excluded. The virus was shown to be resistant to raltegravir in four of the remaining eight patients on raltegravir; in the three cases with available data, the viruses were sensitive to tenofovir and resistant to emtricitabine. Three of the seven patients on efavirenz had efavirenz-resistant virus; in the one case with available data, the efavirenz-resistant virus was also emtricitabine-resistant but susceptible to tenofovir.
Methods
Patients
STARTMRK is a continuing international, double-blind, phase III randomised trial. The design is a non-inferiority comparison. Patients were enrolled from 67 study centres on five continents (Australia, Brazil, Canada, Chile, Colombia, France, Germany, India, Italy, Mexico, Peru, Spain, Thailand, and USA) between Sept 14, 2006, and June 5, 2008, and treated as outpatients. Patients infected with HIV-1 who had not yet received any treatment and were aged at least 18 years were eligible for the study if their vRNA concentration exceeded 5000 copies per mL.
Patients were stratified by baseline vRNA concentration (>50 000 vs ≤50 000 copies per mL) and viral hepatitis co-infection status. Positive hepatitis status was established by detection of hepatitis B surface antigen or hepatitis C RNA (by PCR), or both. Patients were excluded if they had acute or decompensated chronic hepatitis, renal insufficiency (patient needed dialysis, had a serum creatinine concentration of more than twice the upper limit of the normal range, or had an estimated creatinine clearance at screening of ≤30 mL/min as calculated by the Cockcroft-Gault equation), or any medical disorder that could possibly affect the undertaking or interpretation of the study. Patients with chronic hepatitis were eligible if their serum aminotransferase concentrations were less than five times the upper limit of the normal range. Patients infected with HIV strains that had genotypic resistance to tenofovir, emtricitabine, or efavirenz were ineligible; resistance to raltegravir was not tested. Pregnant or breastfeeding women were also excluded.
Ethical approval for the study protocol was provided by the institutional review board or ethical review committee at every study site. All participants provided written informed consent.
Procedures
After stratification, patients were randomly allocated in a 1:1 ratio (double-blind) to receive 400 mg oral raltegravir twice daily or 600 mg oral efavirenz daily, for 96 weeks. Doses of raltegravir were taken 10-14 h apart without regard to food intake, whereas efavirenz was taken on an empty stomach at the hour of sleep. To ensure masking of treatment, patients in the raltegravir group were given placebo tablets identical to efavirenz and vice versa. All participants also took 300 mg tenofovir and 200 mg emtricitabine as a single tablet in the morning with food. Dosing interval adjustment of tenofovir and emtricitabine was recommended for patients with creatinine clearance of 30-49 mL/min.
Once patients had satisfied all eligibility requirements, study site personnel accessed a central interactive voice response system (IVRS) and randomly allocated patients according to a computer-generated randomised allocation schedule to receive raltegravir or efavirenz. Drugs were packaged, assigned unique kit numbers, and aligned with a treatment group in the IVRS database. IVRS selected a kit number associated with the patient's assigned treatment group from the inventory at the study site and instructed the investigator to distribute the kit. Treatment allocation was concealed from investigators, study site personnel, patients, monitors, and central laboratory personnel by use of blinded access codes. Clinical status was assessed at regularly scheduled visits and as needed. Treatment adherence was assessed by review of diaries that were filled out by the study participants and was double-checked by pill counts by site personnel from returned drug bottles.
HIV-1 RNA concentration was measured at a central laboratory by the standard COBAS Amplicor HIV-1 Monitor assay (version 1.5; Roche Diagnostics, Branchburg, NJ, USA) with a low quantification limit of 400 vRNA copies per mL. Up until week 16, the Ultrasensitive Amplicor HIV-1 Monitor assay (version 1.5; Roche Diagnostics) with a low quantification limit of 50 vRNA copies per mL was done on all samples that had less than 400 vRNA copies per mL according to the standard assay. From week 16 onwards, the ultrasensitive assay was used on all samples, followed by the standard assay for samples with more than 50 vRNA copies per mL.
Virological failure could represent either a non-response or a rebound. Non-responders were patients who had 50 vRNA copies per mL or more at either week 24 or the time of premature study discontinuation, without achievement of less than 50 vRNA copies per mL at any stage during the study. Rebounders were patients who after an initial response to treatment, had 50 vRNA copies per mL or more on two consecutive measurements at least 1 week apart.
Emergence of resistance to raltegravir was investigated by genotyping the integrase coding sequence of the virus from patients after virological failure. Consensus amino acid sequences for the integrase gene were compared with pretreatment genotypes. We assessed efavirenz, emtricitabine, and tenofovir resistance at entry of the patient to the study using commercially available assays (Monogram Biosciences, South San Francisco, CA, USA). Because of the nominal limit of the assays, genotyping was done only on viral isolates from patients with more than 400 vRNA copies per mL.
The primary endpoint at week 48 was achievement of less than 50 vRNA copies per mL; the last patient completed the week 48 visit on June 5, 2008. Participants were intended to continue with blinded treatment until week 96. Secondary endpoints at week 48 were the achievement of less than 400 vRNA copies per mL and the change from baseline in CD4 cell count.
Statistical analyses
The primary analysis was per protocol. All randomised patients who were treated with study drug were included in the efficacy and safety analyses of results from baseline to week 48. We postulated that the proportion of patients achieving the primary endpoint with a raltegravir-based regimen would be non-inferior to that with an efavirenz-based regimen. On the basis of 75% of 275 patients in each treatment group achieving a vRNA concentration of less than 50 copies per mL at week 48, the study would have 90% power to show non-inferiority of raltegravir to efavirenz. After adjustment for stratification on the basis of baseline vRNA concentration, a difference in the proportion of patients who responded to treatment of 12% between treatment groups was used to define the non-inferiority margin: raltegravir would be judged non-inferior to efavirenz if the lower bound of the two-sided 95% CI for the proportion of patients who responded in the raltegravir group minus the the efavirenz group was higher than -12%; 95% CIs were calculated according to Miettinen and Nurminen.25 The proportion of patients achieving the secondary efficacy outcome of less than 400 vRNA copies per mL and the change from baseline in CD4 cell count were also recorded for each treatment group.
For calculation of the proportion of patients who achieved less than 50 vRNA copies per mL, the main way to handle missing data was to record all patients who did not complete the study treatment as failures. In this analysis, missing vRNA measurements were imputed as failures, irrespective of the reason for absence, unless the values recorded immediately before and after the missing value were both less than 50 vRNA copies per mL, in which case the absent value was recorded as missing. We also used an observed-failure method to analyse the proportion of patients in baseline subgroups of prognostic indicators who reached the primary endpoint or the proportion who reached immunological endpoints. This analysis, described previously,21, 24 enabled assessment of efficacy without confounding by discontinuations due to intolerability and other reasons unrelated to treatment. Baseline values were carried forward for patients who discontinued because of lack of efficacy; patients who discontinued for other reasons were not recorded in the analyses at subsequent time points. Changes from baseline in CD4 cell counts were summarised during the study by use of the observed-failure method.
Time to virological response was calculated as the time to the first of two consecutive vRNA measurements taken at least 1 week apart in which both had less than 50 vRNA copies per mL. Kaplan-Meier estimates of the time to achieve less than 50 vRNA copies per mL were calculated by treatment group and compared by the log-rank test. All the reported subgroup analyses were predefined in the statistical analysis plan and used the observed-failure approach to missing data. For achievement of the primary endpoint, p values were calculated for non-inferiority (one-sided, α=0·025), and for change in CD4 cell counts, p values were calculated by post-hoc t test (two-sided, α=0·05).
Clinical and laboratory-associated adverse events occurring during the double-blind phase of the study or within 14 days after treatment discontinuation were included in safety analyses. Investigators assessed the presence and nature of a link between every adverse event and the study drug; adverse events were recorded as drug-related if they were judged by the investigator to be definitely, probably, or possibly related to any of the study drugs. The intensity of clinical adverse events was graded by the investigator as mild, moderate, or severe. Severity of laboratory-associated abnormalities was graded according to the Division of AIDS (DAIDS) toxicity guidelines for adults (1992). Information about immune reconstitution was gathered from reports of the adverse-event term of immune reconstitution syndrome. Investigators assessed all other reported adverse events to establish whether the event was due to an immune reconstitution response.
We calculated between-group differences in the proportion of patients with adverse events using the two-tailed Fisher's exact test, and 95% CIs according to Miettinen and Nurminen25 for patients who: (1) had at least one adverse event; (2) had drug-related adverse events; (3) had serious adverse events; (4) had drug-related adverse events that were serious; (5) discontinued study treatment because of an adverse event. With 275 patients in each treatment group, the study had 90% power to show a between-group difference of up to 11·1% with 95% confidence, for adverse events occurring in 20% of patients in either treatment group. If a specific adverse event was not observed in one treatment group, we concluded with 95% confidence that the frequency of this event was 1·3% or lower. We also did a post-hoc analysis of drug-related adverse events with moderate-to-severe intensity.
Adverse event terms were taken from the Medical Dictionary for Regulatory Activities (MedDRA, version 11.0). The frequencies of adverse events were not adjusted for duration of follow-up. A patient was counted only once within a given organ system category, but might be recorded in more than one subcategory or organ system category. To analyse cumulative incidence, we combined several MedDRA terms (depression, nightmare, confusional state, suicidal ideation, nervous system disorder, psychotic disorder, abnormal dreams, suicide attempt, acute psychosis, delirium, depressed level of consciousness, hallucination, auditory hallucination, completed suicide, and major depression) and assessed these as neuropsychiatric symptoms at weeks 8 and 48. After unmasking, we noted that clinically relevant neuropsychiatric symptoms-dizziness, insomnia, somnolence, and impaired concentration-were not included in the list of terms. To assess the robustness of conclusions made from the prespecified list, we analysed the prespecified list combined with assessments of the additional symptoms by investigators unaware of treatment allocation.
To ensure the safety of trial participants and the integrity of the trial, an independent data safety monitoring board (DSMB), consisting of five academicians and one non-voting Merck statistician who was aware of treatment allocation, monitored the trial for evidence of beneficial or adverse effects. The DSMB was to review masked safety and efficacy results every 3 months. The DSMB could request unmasked data and make recommendations for discontinuation of patient enrolment and continuation of the study. Recommendations were given to an oversight committee (members of Merck senior management), which was responsible for the overall scientific direction of the programme and decided whether the study should stop early. All statistical analyses were done with SAS software (version 9.1/8.2).
|
|
| |
| |
|
|
|