| |
HIV, HAART & Brain Damage & Repair
|
| |
| |
Download the PDF
from Jules: this review discusses how despite successful HAART many patients have persistent mild or unrecognized cognitive impairment that could result in serious impairment over time. Perhaps the most important point this article makes is there are potential ways to better characterize & understand the impairment AND POTENTIAL TREATMENTS to repair disease. However, the ACTG, NIAID & the NIH are NOT studying these drugs. It was proposed to the ACTG leadership they study these drugs but the studies were rejected and I don't see anywhere else within the NIH or NIAID a commitment to study these potential therapies. We are NOT adequately addressing CNS/cognitive/neurologic disorders in HIV nor is there a sense of urgency in addressing the aging/HIV related questions that might result in reversing the process apparently contributing to high rates og non-AIDS comorbidities emerging & higher death rates as a result - and these include: senesence, immune activation, and the resulting inflammation. People think inflammation is the problem but inflammation is the result of immune activation & senesence.
Mechanisms of Neuronal Injury and Death in HIV-1 Associated Dementia
"Neuronal damage and loss has been observed in distinct brain regions, including frontal cortex". .... The introduction of highly active antiretroviral therapy ( HAART) has ....Neuronal injury and death induced by HIV-1 infection: Immune- activated and .... disturbance of potential repair mechanisms in the CNS (Fig. ...www.natap.org/2008/HIV/102408_03.htm
HIV and antiretroviral therapy in the brain: neuronal injury and repair
Review
Nature Reviews Neuroscience 8, 33-44 (January 2007) | doi:10.1038/nrn2040
Ronald Ellis1, Dianne Langford2 and Eliezer Masliah3
1. HIV Neurobehavioural Research Center, 150 West Washington St., 2nd Floor, San Diego, California 92103, USA.
2. Department of Pathology, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093, USA.
3. Department of Neurosciences, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093, USA.
Correspondence to: Ronald Ellis1 Email: roellis@ucsd.edu
"it is crucial to identify neural involvement early....Effective cART itself improves the odds that repair will outstrip injury, and by optimizing the CNS penetration of the component medications these odds could be improved even further. However, because not all neurocognitively impaired individuals benefit even from optimal cART, additional therapeutic approaches are needed (see discussion at end and table 1 below)....lithium....Minocycline.....Lexipafant.....memantine.....Early clinical trials suggest that these agents might have some benefits, but they have not been subjected to rigorous scientific evaluation in large, phase III licensing studies."
"The mild degree and fluctuating course of deficits in HIV often obscure their clinical recognition
HIV-associated neurocognitive disorders (HAND)- Neuropsychological impairment is seen in both asymptomatic and symptomatic individuals with HIV..... Most importantly for the future, HAND has been shown to persist, albeit in a less severe form, despite immune reconstitution with cART.....HIV-associated neuropsychological deficits reflect widespread synaptodendritic injury"
"Following cART, although the incidence of HIV-associated neurocognitive disorders has declined, prevalence has not3, 100, presumably due either to longer survival of individuals with milder disorders, incomplete efficacy of cART in the brain, or both."
"The initiation of cART tends to improve overall neurocognitive performance in groups of HIV-infected patients......closer inspection reveals that these improvements are unevenly distributed among patients, with some experiencing remarkable restoration of neurological status whereas others benefit only partially or not at all, remaining mildly or severely impaired. Consistent with the notion that the degree of reversibility of neurocognitive deficits is related to the chronicity and cumulative impact of injury"
"The predominant transition since the introduction of potent combination antiretroviral therapies (cART) has been a replacement of human immunodeficiency virus (HIV)-associated dementia (HAD) and mild neurocognitive disorder (MND) by a milder, chronic asymptomatic neurocognitive impairment (ANI), with which individuals can live for many years1, 142. The impact of a neurocognitive disorder that is severe in a patient who lives for a relatively short period of time might actually be less than the impact of a milder cognitive disorder in an individual who lives for decades. Question marks denote uncertainty as to the longevity of cART-treated individuals."
"Recent work has described various mediators of synaptodendritic repair and restoration after injury in HIV infection....neuroprotective and regenerative factors that have been studied in HIV infection"
table 1
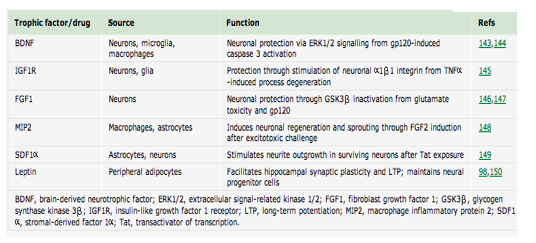
"Understanding the mechanisms through which HIV leads initially to cognitive dysfunction and finding techniques to prevent neural injury or promote recovery is crucial for the development of new treatments for this and other neurodegenerative conditions."
"HIV enters the CNS during the earliest stages of infection.....synaptodendritic degenerative changes in HIV correlate closely to the presence and severity of cognitive impairment....Synaptodendritic injury comprises various structural and chemical changes that affect the 'business ends' of neurons - the synapses at which they communicate and interact with one another....Higher cognitive functions depend on a highly complex synaptodendritic network, and damage to this network results in abnormal output, measured as deficiencies in cognitive skills and behaviour......In one model of HIV infection in the brain, mice with severe combined immunodeficiency (SCID) are genetically engineered to lack specific immune responses, so that it is possible to introduce HIV-infected human monocytes into their brains without evoking a lethal rejection response. These mice show deficits in learning and memory and also show synaptodendritic injuries that are similar to those seen in humans infected with HIV.....Individuals infected with HIV frequently have co-morbidities such as abuse of drugs and alcohol, and infection with viral co-pathogens such as hepatitis C virus (HCV). In neuropathological studies, HIV-infected individuals dying with histories of injection drug use show more activated microglia and diffuse astrogliosis in the white matter of the brain than non-addicted HIV-infected individuals.....HCV infection might increase the likelihood of neurocognitive disorders in HIV-infected individuals. For example, studies have found that HCV infection contributed to the risk of neurocognitive disorder in groups of HIV-infected drug users88, 89, and have also noted that HCV contributed to neurocognitive abnormalities in HIV-infected individuals who had not used drugs. In that HCV is also associated with systemic and possibly CNS immune activation, it is possible that there might be some common immunoneuropathogenic mechanisms leading to an increased likelihood of CNS disease in co-infected individuals."
"The initiation of cART tends to improve overall neurocognitive performance in groups of HIV-infected patients. This includes both patients with clinically recognizable dementia and those with milder impairment. However, closer inspection reveals that these improvements are unevenly distributed among patients, with some experiencing remarkable restoration of neurological status whereas others benefit only partially or not at all, remaining mildly or severely impaired. Consistent with the notion that the degree of reversibility of neurocognitive deficits is related to the chronicity and cumulative impact of injury, patients receiving their first successful cART regimens often show the greatest benefit120. Conversely, successive injuries might reduce the brain's regenerative capacity, correlating with reduced benefits from second, third or fourth attempts at cART. In addition, there is evidence that the degree of penetration of antiretroviral therapy into the CNS (Box 1) influences the extent of neurocognitive improvement120."
Abstract
Approximately 40 million people worldwide are infected with human immunodeficiency virus (HIV). Despite HIV's known propensity to infect the CNS and cause neurological disease, HIV neurocognitive disorders remain under-recognized. Although combination antiretroviral therapy has improved the health of millions of those living with HIV, the penetration into the CNS of many such therapies is limited, and patients' quality of life continues to be diminished by milder, residual neurocognitive impairment. Synaptodendritic neuronal injury is emerging as an important mediator of such deficits in HIV. By carefully selecting specific antiretrovirals and supplementing them with neuroprotective agents, physicians might be able to facilitate innate CNS repair, promoting enhanced synaptodendritic plasticity, neural function and clinical neurological status.
It has long been recognized that the latter stages of infection with human immunodeficiency virus (HIV) often involve severe and disabling dementia. Less well known, however, is that combination antiretroviral therapy (cART) (Box 1), by transforming HIV from a fatal illness into a chronic, manageable condition, has also changed the time course and clinical features of HIV-associated neurocognitive disorders (HAND). Although cART has shown resounding success in treating systemic HIV infection and in partially reversing and preventing severe dementia, milder impairments, which might have a significant adverse impact, remain in a substantial proportion of patients, even before the development of AIDS1, 2, 3. There is no consensus on the relationship between earlier and later neurological effects of HIV. One hypothesis is that the milder neurological dysfunctions and cellular and molecular alterations seen earlier in HIV disease gradually accumulate, leading to the full-blown dementia syndrome seen later. Nevertheless, it is also possible that the mechanisms of disease in these two stages differ qualitatively as well as quantitatively.
The persistence of milder neurocognitive impairment among cART recipients has raised new questions about the causes and treatment of HIV-related brain disorders and about the extent to which neuronal dysfunction is reversible. HIV differs from other neurological conditions such as Alzheimer's disease or Parkinson's disease in that brain function is compromised through indirect mechanisms of neural injury, often without substantial neuronal loss. These non-apoptotic causes of neural dysfunction include axonal disruption and 'pruning' or aberrant sprouting of synaptodendritic connections. Recent theories of HIV neuropathogenesis emphasize that these mechanisms of neurodegeneration lead to compromised neural functioning at a systems level4. Although HIV is a systemic infection, it particularly affects those parts of the brain that govern executive functions, including higher aspects of attention and task-dependent changes in behaviour5, 6.
Although most HIV research has taken place in Western countries, the virus itself is most prevalent and least controlled in resource-limited areas such as sub-Saharan Africa, and East and South Asia7, 8. Findings from studies conducted in Western countries derive mainly from work on HIV-1 clade B infection in Caucasian populations, and therefore might not be generalizable to HIV clades and to other ethnic groups. Additionally, now that antiretroviral therapy is increasingly available internationally, improved management of acute neurological opportunistic infections is critical to short-term survival, which in turn enables patients to initiate long-term antiretroviral therapy. Emerging studies suggest that not only is the prevalence of HIV dementia greater than previously thought, but that the lifespan of those with HIV dementia are shorter9. Importantly, studies also show that, when available, cART is successful at treating HIV dementia in these populations10.
Here, we outline the biology of HIV as it relates to the brain, and the mechanisms by which it causes neurological dysfunction. We then discuss synaptodendritic plasticity and repair and whether HIV-related brain disease can serve as a new model for neural plasticity. Understanding the mechanisms through which HIV leads initially to cognitive dysfunction and finding techniques to prevent neural injury or promote recovery is crucial for the development of new treatments for this and other neurodegenerative conditions.
The biology of HIV
Like all retroviruses, HIV in its genomic, infectious RNA form must be transformed into proviral DNA by a virally encoded enzyme, reverse transcriptase, to become incorporated into the host cell genome. Lentiviruses such as HIV exert pathogenic effects both in the immune system (infecting T lymphocytes and macrophages) and the nervous system. Interestingly, both systems utilize chemical mediators known as chemokines to regulate the complex interactions between cells that are required for normal functioning. HIV can disrupt intercellular communication mediated by chemokines and their receptors, and this seems to be a significant cause of disease in both the immune system and the CNS11 (for additional details on general HIV biology and pathogenesis, see Ref. 12). Among neurotropic viruses, many of which cause disease in fewer than 5% of infected individuals, HIV is distinctly neurovirulent, resulting in neurocognitive impairment in 50% or more of those infected13, 14.
HIV enters the CNS during the earliest stages of infection15.
The HIV surface glycoprotein gp160, which comprises two components, gp120 and gp41, allows the virus to attach to host cell receptors (the CD4 receptor and CXCR4 or CCR5 co-receptor) and to become internalized. CCR5 is the main co-receptor for macrophage-tropic HIV-1 strains, which comprise the vast majority of transmitted HIV and include the predominant strain found in the CNS12. Following infection with HIV, CCR5-bearing macrophages expressing activation markers such as CD16 carry HIV into the brain through a 'Trojan horse' mechanism16. Microglia, the brain's specialized, native immune cells, can also become infected through contact with these trafficking macrophages (for further information on HIV neuropathogenesis, see Ref. 17). As the disease progresses, T-lymphocyte-tropic HIV strains that can use the CXCR4 co-receptor evolve in lymphoid tissues18, 19. These strains are a particularly powerful cause of immunodeficiency (AIDS), which leads to opportunistic diseases, including those that can affect the CNS. The incidence of these life-threatening opportunistic complications has been greatly decreased by the development of effective antiretroviral drugs (Box 1). However, HIV can persist in patients even during such treatment20, probably in resting T cells and CCR5-bearing macrophages, which can harbour multiple alternative forms of viral nucleic acids21, 22.
HIV-associated synaptodendritic injury
Substantial research has been carried out to identify and characterize the neuropathological underpinnings of cognitive impairment in HIV. Although both HIV encephalitis and neuronal apoptosis occur in HIV infection, particularly in association with severe dementia, they do not correspond closely to cognitive impairment23. For example, encephalitis affects some, but not all individuals with HIV, its severity varies greatly, and its presence and extent are not tightly linked to the degree of cognitive impairment24, 25, 26.
By contrast, synaptodendritic degenerative changes in HIV correlate closely to the presence and severity of cognitive impairment27, 28. synaptodendritic injury is a specific type of neuronal injury that is not unique to HIV: it also occurs in diseases as diverse as Alzheimer's disease, Parkinson's disease, schizophrenia, mood disorders and amyotrophic lateral sclerosis29, 30, 31, 32. Synaptodendritic damage can be widespread, as seen in global cerebral ischaemia after cardiopulmonary resuscitation, and this will have a global effect on connectivity. Some diseases show a regional predilection: for example, in Borna disease, Alzheimer's disease and some forms of epilepsy, synaptodendritic injury is characteristically greatest in the hippocampal dentate gyrus33. In HIV, although synaptodendritic injury is distributed widely throughout the brain, some regions of the forebrain show selective vulnerability (see below).
Synaptodendritic injury comprises various structural and chemical changes that affect the 'business ends' of neurons - the synapses at which they communicate and interact with one another. Structurally, the normal synaptodendritic network is complex and highly branching, whereas the injured network is pruned, with varying degrees of loss or attenuation of complexity (Fig. 1). Injury or insult to the dendritic arbor is also seen as retraction of dendritic spines, dendritic beading and aberrant sprouting, all of which can occur without neuronal death. The result of these pathological processes is disruption or loss of normal synaptodendritic communication and axoplasmic flow. Higher cognitive functions depend on a highly complex synaptodendritic network, and damage to this network results in abnormal output, measured as deficiencies in cognitive skills and behaviour.
In preserved tissue, synaptodendritic injury is demonstrated by immunostaining with antibodies to presynaptic synaptophysin (SYP) and postsynaptic microtubule-associated protein 2 (MAP2)28 (Fig. 1a,b). MAP2 is expressed in neuronal cell bodies and dendrites. It is involved in microtubule assembly and interacts with microtubules, neurofilaments and actin filaments. As such, it serves as a marker of the integrity of the neuronal dendritic network. In HIV-infected individuals, the degree of neurocognitive impairment is strongly related to loss of immunostaining for SYP and MAP2. This technique has provided evidence that the striatum (made up of the putamen and caudate nucleus) and the hippocampus are particularly affected34, 35. This regional vulnerability parallels the higher burden of HIV proteins and viral RNA in the striatum and white matter connecting the striatum to the prefrontal cortex36, 37. Similar changes are seen in animal models of HIV-related neuropathology, which are discussed below. Moreover, the extent of synaptodendritic injury in these regions correlates with the degree of neuropsychological impairment in humans.
Recently, in vitro techniques have been developed to portray synaptodendritic damage dynamically38. Neurons from embryonic rats are transfected with genes for red or yellow fluorescent proteins (mRFP or EYFP) which, when subsequently expressed, label the neuronal cell bodies and dendritic and axonal processes. Serial photographs of the cells' morphologies are taken at various times after exposure to drugs or toxins. Such techniques have been used to characterize dendritic beading (Fig. 2). Neurons that undergo dendritic beading lose their capacity for long-term potentiation (LTP)39, a central process in memory formation. Moreover, certain types of LTP underlie regeneration, plasticity and recovery of function after neuronal injuries and degeneration. Importantly, both the structural and functional changes that occur with dendritic beading can be reversible, such that normal anatomy and function can be restored. This raises the possibility that therapeutic interventions could be designed to assist the repair of damaged synaptodendritic connections, as discussed below.
It is difficult to assess synaptodendritic injury during life. One approach is to measure synaptodendritic proteins released from damaged neurons into the extracellular space, which can be detected in the cerebrospinal fluid (CSF) or blood. Elevated CSF neurofilament protein (NFL) concentrations, for example, are thought to reflect injury to myelinated axons. CSF NFL levels are increased both in the context of HIV dementia40, 41 and also after the interruption of cART, which results in a marked rebound of HIV replication. Levels of NFL in the CSF are also increased in Alzheimer's disease, multiple sclerosis and other conditions42. However, it is not known whether increased NFL can result from neuronal injury, or whether it requires cell death, and there are no data on the dynamic relationship between changes in neurological status in patients with HIV and changes in NFL levels.
An alternative approach is to look at the brain as a whole by using neuroimaging techniques. Macroscopically, loss of dendritic and synaptic structures in HIV can reduce the overall volume of the neuropil and the white matter. These losses summate to gross structural atrophy that can be visualized by brain computed tomography and MRI. Careful radiological-neuropathological correlation studies have shown that white matter loss and abnormal white matter signal are closely correlated with the loss of MAP2 immunostaining, particularly in the presence of HIV encephalitis43. Worsening synaptodendritic abnormalities and MRI white matter changes correlate with worsening cognitive impairment27, 44.
The molecular and cellular correlates of neurocognitive dysfunction in early- and mid-stage HIV disease have been difficult to delineate, as human CNS tissues rarely become available for study until patients die with advanced AIDS. Nevertheless, by evaluating rhesus monkeys infected with simian immunodeficiency virus (SIV), a virus with close genomic and pathogenic similarities to HIV, it has been possible to determine correlates of earlier stage HIV CNS disease45. Infected animals at such earlier stages show cognitive, behavioural and electrophysiological abnormalities, and both SIV and infiltrating CD8+ lymphocytes are found in their brain tissue46. Additionally, immune response genes such as CCL5 are upregulated in the brain throughout the course of disease46.
Studies in experimental animal models as well as observations in the brains of patients with AIDS indicate that neuronal damage might initially occur in synapses and dendrites and then spread to the rest of the neuron, thereby activating pathways leading to cell death by apoptosis27, 28, 47. Supporting this possibility, the brains of patients with HIV encephalitis show evidence of DNA fragmentation in neurons, glia and endothelial cells, as determined by the terminal deoxynucleotidyl transferase labelling (TUNEL) assay48, 49. Moreover, in HIV encephalitis, caspase 3 - a protease enzyme that is involved in triggering or mediating apoptotic neuronal death - is activated and pro-apoptotic gene expression is upregulated47, 50. The mechanisms involved in neuronal and synaptic degeneration in patients with HIV encephalitis are complex, but most evidence supports the contention that infected and/or activated microglia and macrophage cells release toxic factors such as viral products, excitotoxins, and/or cytokines and chemokines that in turn damage neurons through multiple mechanisms51, 52, 53. These factors might also activate astrocytes which then produce chemokines and cytokines that affect neuronal function54, 55. Such neurotoxic factors are likely to affect a diverse range of neuronal populations in the most vulnerable areas of the CNS, including the frontal cortex, basal ganglia, hippocampus and white matter.
Certain types of neuron in these vulnerable regions are particularly susceptible to synaptodendritic and apoptotic damage as a result of HIV infection. For example, excitatory pyramidal neurons in the neocortex are extensively damaged, as are GABA (gamma-aminobutyric acid)-producing, calbindin-immunoreactive interneurons in the neocortex and hippocampus. Calbindin is a 28 kDa calcium-binding protein that is expressed at high concentrations by inhibitory neurons, but not by pyramidal excitatory neurons56. Calbindin buffers intracellular calcium currents and has been shown to be protective against neuronal excitotoxicity57. In the hippocampus, calbindin-immunoreactive inhibitory neurons innervate sites near the distal dendrites of other neurons and are believed to regulate the firing rate of pyramidal excitatory neurons58, 59, 60. Calbindin deficiency in the hippocampal dentate gyrus impairs LTP61, indicating that calbindin helps to regulate LTP during memory formation62. Activity of neocortical calbindin-immunoreactive interneurons is also important in modulating bidirectional neuronal responses in the prefrontal cortex that regulate cognitive functions such as working memory63. In HIV encephalitis, calbindin-immunoreactive double bouquet interneurons are preferentially affected. Together, these pathophysiological mechanisms would be predicted to cause alterations in intrinsic hippocampal and frontostriatal circuits, resulting in deficits in learning and executive functions, as described below.
Mechanisms of HIV-associated injury
The effect of chronic infection with HIV on molecular pathways in the CNS, resulting in both synaptodendritic injury and apoptosis, is often described as involving two overarching aspects: viral factors and host factors. Viral factors, such as proteins encoded by the viral genome, derive more or less directly from the virus itself. Host factors derive indirectly or secondarily from infection, and propagate as a cascade of molecular events that damage uninfected cells, most importantly neurons themselves. Host factors can differ between individuals - for example, increased susceptibility to neurodegeneration in HIV is associated with host genetic variations that might account for differential susceptibility to dementia between different individuals infected with the same virus64. This is similar to the situation with cerebral malaria, where homozygotes for the tumour neurosis factor 2 (TNF2) allele, a variant of the TNFalpha gene promoter region, show a markedly increased risk of death and severe neurological sequelae65.
Neurotoxicity of viral factors.
The HIV genome codes for various proteins that, apart from their roles in the viral life cycle, can damage neuronal cells and interfere with the function of the CNS. For example, the virally encoded HIV envelope protein, gp120, is necessary for infectivity, but also interacts with cellular receptors to alter glutamate pathway signalling and induce cytokine production that can injure neurons and affect the activation state of microglia and astrocytes. Figure 3 shows components of the viral genome and lists some of the ways in which they can contribute to neuronal injury.
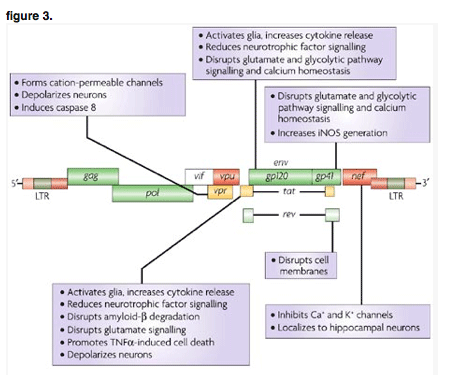
The human immunodeficiency virus (HIV) provirus is approximately 9.8 kb in length and encodes at least nine proteins. The genes encode proteins that can be divided into three classes: structural - Gag, Pol (polymerase) and Env (envelope); regulatory - Tat (transactivator of transcription) and Rev (regulatory for expression of viral proteins); and accessory - Vpu (viral protein u), Vpr (viral protein r), Vif (viral infectivity factor) and Nef (negative factor). HIV proteins released from infected cells can have direct effects on cells of the CNS. iNOS, inducible nitric oxide synthase; LTR, long terminal repeat; gp, glycoprotein, TNFalpha, tumour necrosis factor-alpha.
Animal and cell culture models have been used to characterize viral factor-mediated synaptodendritic injury and elucidate its mechanisms. For example, the dendritic alterations seen in the brains of cognitively impaired HIV-infected individuals that come to autopsy (Fig. 1) can be replicated in cultures of human nervous tissue exposed to gp120 (Ref. 66). One molecular mechanism by which gp120 might induce these changes is through glutamate-mediated excitotoxicity, which can initiate caspase cascades67. Caspases are upregulated both in neuronal cultures exposed to gp120 and in the brains of cognitively impaired individuals with HIV infection. Garden et al.47 showed that the elevations of caspase 3 activity that occurred in gp120-exposed cultures were confined to dendrites and the neuronal soma, and were not accompanied by apoptotic changes in the cell nuclei. In addition, specific inhibitors of caspase 3 could reverse these degenerative dendritic changes in vitro. These findings indicate that caspases might be involved in both non-apoptotic neurodegeneration and in normal synaptic plasticity. This opens potential avenues for therapeutic interventions designed to ameliorate synaptodendritic injury induced by HIV and other diseases.
Transgenic mice producing HIV-1 gp120 in their brains show neuropathological features that are similar to those found in the brains of neurocognitively impaired, HIV-infected humans68. In addition to synaptodendritic injury, these changes include reactive astrocytosis, increased number and activation of microglia, and loss of large pyramidal neurons69. gp120 mediates such damage, at least in part, by overactivation of glutamate receptors, leading to increased extracellular glutamate that can cause neuronal excitotoxic damage. In support of this, nanomolar concentrations of gp120 have been reported to interact with the glycine binding site of the NMDA (N-methyl-D-aspartate) receptor70.
In one model of HIV infection in the brain, mice with severe combined immunodeficiency (SCID) are genetically engineered to lack specific immune responses, so that it is possible to introduce HIV-infected human monocytes into their brains without evoking a lethal rejection response71. These mice show deficits in learning and memory, for example in a Morris water maze72, and also show synaptodendritic injuries that are similar to those seen in humans infected with HIV73. They have significant astrogliosis and microgliosis, similar to the levels of gliosis seen in HIV-infected humans, and they show reductions in MAP2 immunostaining of dendrites that also replicate the changes seen in the brain tissue of cognitively impaired patients with HIV. Alterations in dendritic morphology and neuronal physiology are more pronounced than actual neuronal loss (apoptosis), both in HIV-infected SCID mice and in humans49, 72.
Another viral protein that can cause neuronal injury is transactivator of transcription (Tat), which can be produced by astrocytes74. When astrocytes are infected with HIV, the infection does not produce whole virions, but instead generates Tat. In experiments in which tat-expressing astrocytes were injected into the rat dentate gyrus, Tat was taken up by granule cells and transported along neuronal pathways to the CA3 region of the hippocampus, where it caused glial cell activation and neurotoxicity34. Tat can also cause mitochondrial dysfunction, dendritic loss and cell death in neurons at concentrations lower than those needed to support viral replication75.
Host factors in synaptodendritic injury and neuronal apoptosis.
In addition to the toxic effects of HIV itself, secondary effects of HIV infection on the immune system can amplify nervous system damage. So, many of the host factors relevant to HIV CNS injury are chemical mediators of inflammation and immunity - cytokines and chemokines. The upregulation of chemokines and chemokine receptors in the CNS is an emerging theme in the study of neurodegenerative diseases76. For example, chemokines and their receptors are abundantly expressed in multiple sclerosis lesions, cerebral infarcts and reactive glia around senile plaques in Alzheimer's disease, and they are believed to contribute directly and/or indirectly to neurodegeneration (for a review, see Ref. 76). This effect is often referred to as chronic neuroinflammation.
Inflammation can be a more or less episodic event that resolves and leaves in its wake a cascade of neurodegeneration that carries on for some time. Cytokines that traffic into the CNS from the peripheral circulation are produced by monocytes and macrophages, as well as by astrocytes and microglia activated or infected by HIV. Chemokines represent a subset of cytokines with chemoattractant properties, and are particularly important in HIV-related pathology. Chemokine receptors are distributed throughout the brain on microglia, astrocytes, oligodendrocytes and neurons. Abnormal activation of cytokine and chemokine receptors in the context of HIV infection results in dendritic beading and loss of dendritic spines. These changes are accompanied by failure of LTP, which might underlie impaired learning and memory77. Of note with regard to potential therapies is that these ultrastructural and functional alterations are at least partially reversible (Fig. 2). Therefore, it might be possible to design treatments that could accelerate restoration of neural alterations arising from abnormal activation of cytokine or chemokine receptors.
The dividing line between viral and host-mediated neurotoxicity in HIV infection is neither precise nor rigid. So-called 'viral' toxicity is sometimes amplified or provoked by host-derived cofactors. For example, in mixed neuronal and glial cerebrocortical cultures, HIV gp120-induced injury requires the release of microglial toxins11, 78, 79, 80. As a result, effective treatment of HIV CNS disease might require both cART and so-called 'adjunctive' therapies that include neuroprotective and neuro-regenerative agents.
Cofactors in HIV nervous system injury.
Individuals infected with HIV frequently have co-morbidities such as abuse of drugs and alcohol, and infection with viral co-pathogens such as hepatitis C virus (HCV). In neuropathological studies, HIV-infected individuals dying with histories of injection drug use show more activated microglia and diffuse astrogliosis in the white matter of the brain than non-addicted HIV-infected individuals81, 82. Langford et al.83 found that those dying with HIV and a history of methamphetamine addiction had a greater loss of calbindin-immunostaining interneurons than in those with either condition alone. This study also found more severe microglial reactions in those with histories of methamphetamine dependence, suggesting a methamphetamine-associated immunological stimulation. In support of this possibility, monocyte chemoattractant protein 1 (MCP1) is selectively elevated in the CSF of methamphetamine abusers84. Consistent with these observations, brain samples of individuals dying with methamphetamine dependence show upregulation of interferon-inducible genes85, providing a potential mechanism for methamphetamine-induced inflammation in the CNS. A recent study found that the degree of dendritic simplification was associated with both methamphetamine and HIV, with an indication of an additive effect in the presence of both factors86. On the clinical side, another study showed that HIV-infected methamphetamine users were more likely to have various neurocognitive impairments than patients with either risk factor alone, again suggesting an additive effect87.
Although it is beyond the scope of this review to consider infectious co-morbidities exhaustively, mention should be made of the emerging recognition that HCV infection might increase the likelihood of neurocognitive disorders in HIV-infected individuals. For example, studies have found that HCV infection contributed to the risk of neurocognitive disorder in groups of HIV-infected drug users88, 89, and have also noted that HCV contributed to neurocognitive abnormalities in HIV-infected individuals who had not used drugs. In that HCV is also associated with systemic and possibly CNS immune activation, it is possible that there might be some common immunoneuropathogenic mechanisms leading to an increased likelihood of CNS disease in co-infected individuals.
Mechanisms of repair
In the healthy brain, synapses undergo continuous remodelling, which is sometimes referred to as plasticity - a term also used in the context of restoration of function after brain injury. Neuronal plasticity can include increased dendritic branching, augmentation of axonal collaterals, the generation of new synaptic connections and activity-dependent modification of existing synapses90. More recently, evidence has emerged that neurogenesis can also contribute to the ability of the diseased nervous system to restore some functions after injury91, 92. The plasticity of synaptodendritic structures in response to injury can be shown dynamically using in vitro models. For example, in a simple neuronal culture system (Fig. 2), neurons treated with supraphysiological concentrations of potassium chloride undergo depolarization, release neurotransmitters and rapidly develop dendritic beading93. This beading reverses once the potassium chloride is removed, with full, spontaneous recovery after 5-10 minutes. Exposure of such neurons to the cytokine platelet activating factor (PAF), an immune mediator implicated in HIV-associated neuronal injury, increases their vulnerability to dendritic alterations, so that lower doses of toxin are required to reproduce the alterations. The same group of investigators93 showed dendritic beading in hippocampal slice preparations, a more complex model that is perhaps closer to the living brain. Dendritic beading in this model showed different temporal and spatial features from the simple neuronal culture system. High-frequency stimulation of Schaffer collaterals in hippocampal slices led, after a delay, to dendritic beading in hippocampal neurons that lasted from minutes to hours, and was dependent on caspase activity. This type of dendritic beading disrupted discrete regions of dendrites but left most of the dendritic arbor intact. Again, spontaneous recovery of normal dendritic morphology occurred if the stimulus was removed.
Recent work has described various mediators of synaptodendritic repair and restoration after injury in HIV infection. These mediators take advantage of a tightly linked crosstalk among neurons, glia, oligodendrocytes and cerebral endothelial cells that is essential for synaptodendritic plasticity during pathological conditions in the brain17, 94, 95. Table 1 lists some of the neuroprotective and regenerative factors that have been studied in HIV infection, emphasizing the highly interactive nature of communications among CNS cells under physiological and pathological conditions. For example, in response to injury or challenge, cells of the CNS, including neurons, produce cytokines and trophic factors such as brain derived neurotrophic factor (BDNF), insulin-like growth factor 1 (IGF1), acidic fibroblast growth factor (aFGF, FGF1), macrophage inflammatory protein 2 (MIP2), stromal-derived factor 1alpha (SDF1alpha) and leptin96, 97, 98. One way in which HIV might contribute to nervous system disease is by interfering with these trophic and neuroprotective pathways. Viral proteins or virally induced immune changes can intersect with the pathways involved in trophic factor gene expression, protein production or signalling cascades. Subsequent reductions in trophic factors and signal transduction can increase neurons' vulnerability to injury and limit their ability to undergo repair. Treatment avenues could potentially take advantage of these mechanisms by therapeutically manipulating the expression of trophic and protective factors (see below).
Clinical impact of HIV in the CNS.
Neurodegeneration and synaptodendritic injury in HIV-infected individuals result in the clinical syndromes of HAND. Neuropsychological testing of large numbers of HIV-infected individuals has shown that both in the pre-cART and post-cART eras, up to 50% of patients experience HAND44, 99 (Fig. 4). Before cART, severe and disabling dementia (HIV-associated dementia, HAD) affected approx20% of patients with advanced HIV disease, usually shortly before death. Following cART, although the incidence of HIV-associated neurocognitive disorders has declined, prevalence has not3, 100, presumably due either to longer survival of individuals with milder disorders, incomplete efficacy of cART in the brain, or both101.
When present, neurocognitive disorders can range in severity from subtle deficits to incapacitating dementia. Although impairment tends to be milder early in the course of infection, increasing in prevalence and severity with advancing disease99, 102, varying degrees of impairment occur at all stages. There is evidence that higher viral loads in the CSF are associated with the subsequent development of HAND103. Three diagnostic terms are in common use. Asymptomatic neurocognitive impairment (ANI) refers to the presence of mild neurocognitive deficits, established by neuropsychological testing, which have not progressed to a point at which the individual is aware of substantial interference in everyday functioning. Mild neurocognitive disorder (MND; also known as minor cognitive motor disorder, MCMD) indicates the presence of several cognitive deficits that are mild to moderate in degree and which interfere to a modest extent with everyday functioning. Individuals with MND can typically continue to work and carry out other activities of everyday living, albeit in a less efficient manner. HAD describes individuals with multiple moderate to severe cognitive impairments which markedly disturb everyday functioning, and almost always render the person incapable of living independently in the community.
Most HIV-infected individuals who suffer from neurocognitive disorders have ANI or MND, and the presence of these conditions seems to reflect underlying neurobiological changes. For example, there is a strong monotonic relationship between severity of cognitive impairment before death and the degree of loss of dendritic arbor at autopsy27, 28. Compared with cognitively intact HIV-infected individuals, those with HAND have a lower quality of life104, are more likely to be unemployed105, 106, 107 and have greater difficulty with vocational functioning99, medication management6, driving108 and other activities of daily living. Neuropsychological impairment is seen in both asymptomatic and symptomatic individuals with HIV. Among individuals with known dates of initial HIV infection, new-onset neuropsychological impairment preceded opportunistic infections in 12% of cases109. Most importantly for the future, HAND has been shown to persist, albeit in a less severe form, despite immune reconstitution with cART3, 110.
HIV-associated neuropsychological deficits reflect widespread synaptodendritic injury, particularly involving fronto-striato-thalamo-cortical circuits (FSTC circuits). FSTC circuits reciprocally link the prefrontal cortex to the striatum, to other basal ganglia structures such as the globus pallidus, to the thalamus and back to the prefrontal cortex111. As noted earlier, indicators of white matter damage and markers of synaptodendritic injury such as MAP2 and SYP reflect injury to these interconnected systems43. By contrast, cognitive functions mediated by posterior neocortical and temporolimbic systems, such as object naming, visual perception and memory consolidation, are typically spared in HIV infection107.
The prototypical cognitive profile of HIV-1 infection involves deficits in cognitive domains that are highly dependent on these frontostriatal circuits. Frequently observed executive dysfunctions include disturbance in abstraction44, 99, 112, speeded information processing99, verbal fluency4, decision making113 and various aspects of attention114, 115. There is also memory disturbance, the features of which more closely resemble those seen in Parkinson's disease and Huntington's disease rather than cortical dementias such as Alzheimer's disease. The primary difficulties tend to be in components of learning new information such as maintaining information in working memory116, 117 and encoding118, rather than accelerated forgetting.
Recent observations also indicate that another aspect of memory, termed prospective memory, might be affected by HIV119. Prospective memory is a unique and dissociable form of episodic memory involving the ability to execute a future intention, or 'remembering to remember'. Prospective memory is essential for forming, monitoring and executing plans of behaviour in the context of ongoing distractions. Everyday examples include remembering to take a medication at specific times or remembering to attend a scheduled medical appointment. Carey et al. showed that HIV-infected individuals were impaired in time- and event-based prospective memory and showed more frequent 24 hour delay failures in prospective memory compared with HIV-uninfected controls.
Time course of HIV neurocognitive disorders
As illustrated in Fig. 5, the longitudinal course of HIV neurocognitive disorders can be variable. Of the approx50% of HIV-infected individuals who develop cognitive impairment, roughly one-fifth show a fluctuating course and the remainder have varying patterns such as complete recovery or, occasionally, progressive worsening. The mild degree and fluctuating course of deficits in HIV often obscure their clinical recognition.
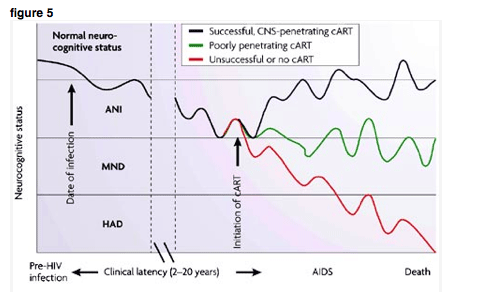
The progression of cognitive impairment in patients with human immunodeficiency virus (HIV) is unpredictable, waxing and waning over time, but does vary depending on treatment. The figure illustrates a hypothetical course of neurocognitive disorder in three patients with HIV. The red line represents an individual taking no or ineffective antiretroviral therapy. Both viral and host factors contribute to cumulative injury and a reduction in the capacity for repair of the synaptodendritic network. The green line represents an individual who has undergone successful systemic treatment of HIV, but with only partial CNS efficacy due to limited penetration of antiretroviral drugs into brain tissue. The blue line represents an individual who has undergone successful treatment of both systemic and CNS HIV infection by using combined antiretroviral therapy (cART) with good CNS penetration. Normal neurocognitive status, no neurocognitive impairment. Asymptomatic neurocognitive impairment (ANI), mild neurocognitive impairment, not clearly affecting daily activities. Mild neurocognitive disorder (MND; also known as minor cognitive motor disorder, MCMD), mild-moderate neurocognitive impairment with early functional loss. HIV associated dementia (HAD), moderate-severe neurocognitive impairment with obvious deficits in activities of daily living. Unlike in many neurodegenerative disorders such as Alzheimer's disease, the various degrees of HIV neurocognitive dysfunction depicted here are not true stages, in that more severe degrees of impairment do not necessarily follow milder ones in a predictable order. In fact, some investigators believe that these types of neurocognitive dysfunction are qualitatively as well as quantitatively different.
Above, we described various intricate mechanisms underlying neuronal injury and repair in HIV. If HIV represents a disease model in which, in addition to a cumulative history of injury, there is a dynamic, fragile balance between injury and repair, it is easy to imagine how disturbances in this delicate balance might result in waxing and waning of cognitive function over time. A snapshot at one point in time of neurological status or of indicators reflecting injury and repair might provide a misleading portrait of the overall time course. In a chronic disease such as HIV, the temporal course of CNS dysfunction becomes particularly important to consider. Even before the advent of effective antiretroviral therapy, the time course of HIV - 5-20 years from infection to death - was prolonged relative to many other diseases. With the advent of cART, survival has been extended even further, amplifying the importance of the temporal dimension.
The initiation of cART tends to improve overall neurocognitive performance in groups of HIV-infected patients. This includes both patients with clinically recognizable dementia and those with milder impairment. However, closer inspection reveals that these improvements are unevenly distributed among patients, with some experiencing remarkable restoration of neurological status whereas others benefit only partially or not at all, remaining mildly or severely impaired. Consistent with the notion that the degree of reversibility of neurocognitive deficits is related to the chronicity and cumulative impact of injury, patients receiving their first successful cART regimens often show the greatest benefit120. Conversely, successive injuries might reduce the brain's regenerative capacity, correlating with reduced benefits from second, third or fourth attempts at cART. In addition, there is evidence that the degree of penetration of antiretroviral therapy into the CNS (Box 1) influences the extent of neurocognitive improvement120.
Potential avenues for diagnosis and treatment
Early detection of HIV-related neurological deficits.
Given the evidence that cumulative injury over time can result in a non-reversible component of HAND, it is crucial to identify neural involvement early. Sensitive neuropsychological testing, using measures optimized for the detection of HIV-related deficits, and interpreted using appropriate normative comparisons represents the gold standard for detection of HAND. However, other technologies show promise for early detection of synaptodendritic injury, perhaps even before it affects neurocognitive performance. For example, recent findings from brain functional imaging studies support the notion that, before a breakdown in the accuracy or speed of cognitive performance, the HIV-infected brain recruits or activates more widespread regions to accomplish the same level of performance121.
Blood-oxygen-level-dependent (BOLD) functional MRI (fMRI) has been used to evaluate attention and memory in HIV patients114, 121, 122, 123. BOLD maps associated with a simple cognitive task have shown decreased activation in areas commonly associated with attention, with increased activation in adjacent and contralateral brain regions. These results might reflect reduced efficiency in typical attentional networks and the recruitment of additional neural areas to compensate for the disruption in normal synaptodendritic activity. Another MRI technique, arterial spin labelling (ASL), can be used to study baseline cerebral blood flow. Measurements of cerebral blood flow in the caudate nucleus of HIV patients on stable cART with varying levels of cognitive performance from normal to HAD show a significant decrease in resting perfusion that correlates with increasing cognitive impairment124. Non-invasive imaging techniques might provide opportunities to identify early synaptodendritic injury, and to monitor the effects of neuroprotective cART and other interventions.
Treatment options.
As the mechanisms that underlie synaptodendritic injury and repair are dissected at a molecular level, opportunities arise to design interventions that might facilitate repair and restoration of synaptodendritic integrity. Effective cART itself improves the odds that repair will outstrip injury, and by optimizing the CNS penetration of the component medications these odds could be improved even further. However, because not all neurocognitively impaired individuals benefit even from optimal cART, additional therapeutic approaches are needed. For example, trophic or protective factors could be supplied by gene therapy or other means to reverse existing injury or to arrest further damage, as has been tried in Alzheimer's disease125.
Other approaches involve the administration of pharmacological agents that facilitate endogenous repair or trophic mechanisms. For example, lithium, which is used to treat bipolar disorder, also modulates the expression of the pro-apoptotic kinase glycogen synthase kinase 3beta. In vitro, lithium prevents the induction of dendritic spine loss and simplification by HIV-1 gp120 (Ref. 126), indicating that it might be useful in anti-HIV therapy. In individuals with HAND, lithium administration was associated with improved neurocognitive performance in a single-arm, 12 week study127. Minocycline is an antibiotic that has secondary anti-inflammatory and neuroprotective effects linked to suppression of p38 mitogen-activated protein kinase. In rhesus macaques infected with SIV, minocycline treatment reduced the activation of macrophages and microglia and decreased the influx of cytotoxic lymphocytes into brain tissue, reducing the severity of encephalitis, suppressing viral load and dampening the expression of CNS inflammatory markers128. Minocycline trials in humans are being planned. Lexipafant protects neurons from calcium- and caspase-induced dendritic beading and loss of LTP mediated by PAF93, and memantine, a non-competitive antagonist of the NMDA receptor, prevents Tat- and gp120-induced intracellular calcium increases and glutamate toxicity129, 130, 131, 132, and has been applied in human trials. Early clinical trials suggest that these agents might have some benefits, but they have not been subjected to rigorous scientific evaluation in large, phase III licensing studies.
Summary and conclusions
HIV-associated neuropathology results from multiple direct and indirect injuries to the synaptodendritic network mediated by both viral and host factors. The consequences for affected individuals include neurocognitive disorders ranging from mildly impaired neuropsychological test performance to disabling dementia. Not only does the degree of cognitive impairment vary among patients, but the severity of impairment at any given time in a single patient can also fluctuate. We propose that these perturbations reflect disturbances in the dynamic balance between synaptodendritic injury and repair. So, virus- and host-derived toxic factors that are active during HIV infection are counteracted by host trophic factor production and neuronal plasticity. The introduction of cART has reduced the prevalence of severe dementia, but patients have not tended to recover full cognitive function, resulting in an overall increase in the proportions of individuals with milder degrees of HIV-related neurocognitive disorders. This means that, in the cART era, neurocognitive status for many has reached a suboptimal plateau5, 111, 120, 122, 133, 134, 135, 136, 137, 138.
In vitro evidence of dynamic shifts in the balance of synaptodendritic injury and repair over time are consistent with the multiphasic clinical profile of HIV neurocognitive disorders. These various phases seem to reflect different molecular and ultrastructural mechanisms with different time courses. Being able to identify these phases might help clinicians to target therapies at different stages of recovery to maximize clinical benefit. In this way, HIV could serve as a model for other specific conditions that involve chronic synaptodendritic injury, such as other infectious or inflammatory diseases including systemic lupus erythematosus, multiple sclerosis and neurological Lyme disease and HCV infection. Ongoing and future studies are addressing the early identification of HIV-related neural injury using molecular markers and neuroimaging techniques, and antiretroviral therapies that specifically target CNS HIV infection - as distinct from systemic infection - as well as adjunctive therapies that reduce host factor-mediated damage or support mechanisms of neural repair.
|
|
| |
| |
|
|
|