 |
|
|
| |
Comparable Safety and Efficacy With Once-daily (QD) Versus Twice-daily (BID) Dosing of Lopinavir/ritonavir (LPV/r) Tablets With Emtricitabine (FTC) + Tenofovir DF (TDF) in Antiretroviral (ARV)-naïve, HIV-1-infected Subjects:
96-Week Results of the Randomized Trial M05-730
|
| |
| |
Reported by Jules Levin
5th IAS Conference on HIV Pathogenesis, Treatment, and Prevention · 19-22 July 2009 · Cape Town, South Africa
Juan Gonzalez Garcia1, Daniel Cohen2, Margaret Johnson3, Louis Sloan4, Linda Fredrick2, Christian Naylor2, Barbara da Silva2, Barry Bernstein2
1Hospital Universitario La Paz, Madrid, Spain; 2Abbott Laboratories, Abbott Park, IL USA; 3Royal Free Hospital NHS Trust, London, UK; 4North Texas Infectious Disease Consultants, Dallas, TX USA
Presenting author: Daniel Cohen, M.D., Abbott Laboratories, 200 Abbott Park Rd., Abbott Park, IL 60064 USA; Tel: +1-847-938-1494; Fax: +1-847-938-3711; E-mail: daniel.cohen@abbott.com
Author Discussion
This is the longest and largest study comparing QD and BID LPV/r using the tablet formulation.
This comparison of QD versus BID LPV/r dosing in antiretroviral-naïve, HIV-1-infected subjects demonstrated that through 96 weeks, the efficacy of LPV/r QD
continued to meet the protocol-defined criteria for non-inferiority compared to BID. This finding is consistent with previous studies as well as the 48-week results from Study M05-730.1-3
These results are in accordance with a meta-analysis that revealed no effect of LPV/r dose frequency on efficacy in difficult-to-treat subjects with high baseline
HIV-1 RNA or low baseline CD4+ T-cell counts.5
Importantly, the overall emergence of post-baseline resistance mutations was rare and was not significantly different with QD or BID dosing. There was no evidence of protease inhibitor-associated resistance in either dosing group, a
finding consistent with the previous reports of the high genetic barrier observed with LPV/r therapy.
Intermittent viremia occurred with a similar low frequency in QD and BID treated subjects, and was not associated with the development of LPV- or TDF associated mutations in any subject.
Through 96 weeks of treatment, no clinically significant differences in the safety or tolerability of QD compared with BID LPV/r were detected, with similar rates of
moderate-to-severe diarrhea and a low frequency of Grade 3+ laboratory abnormalities.
As previously noted with LPV/r therapy, increases in some lipid parameters were detected. These changes were largely unaffected by LPV/r dose frequency, with
the exception of triglycerides and non-HDL cholesterol, which showed slightly smaller mean increases, and the LDL:HDL ratio, which had a marginally greater mean decrease, in QD subjects compared to BID. The clinical significance of these fi ndings is unknown.
Author Conclusions
QD LPV/r shows durable antiviral activity in antiretroviral-naïve, HIV-1-infected subjects through 96 weeks, with similar efficacy compared to BID.
Comparable efficacy of QD compared to BID LPV/r was also observed in subgroup analyses, regardless of baseline CD4+ T cell count or HIV-1 RNA level.
LPV/r tablets dosed QD and BID show similar safety and tolerability, with low rates of treatment discontinuation due to adverse events.
QD and BID LPV/r appear to have a comparable high barrier to resistance based on the low rate of emergence of resistance through 96 weeks.
Background
Lopinavir/ritonavir is a coformulation of the HIV-1 protease inhibitor lopinavir with low-dose ritonavir, which acts as a pharmacokinetic enhancer to increase
lopinavir bioavailability, and is indicated globally for treatment of HIV-1 infection when used in combination with other antiretroviral agents.
Initial studies using the soft gel capsule (SGC) formulation suggested that QD LPV/r offers non-inferior efficacy compared to BID dosing in antiretroviral-naïve
subjects.1, 2
The LPV/r tablet formulation offers the advantages of a reduced pill burden (4 tablets daily compared to 6 SGCs), lack of a food effect, and no need for
refrigeration.
Study M05-730 was a large, randomized, open label study designed to evaluate QD versus BID dosing with the LPV/r tablet formulation in antiretroviral-naïve
subjects through 96 weeks of treatment. It also compared tolerability between the tablet and SGC formulation through the first 8 weeks.
The previously-presented 48-week primary efficacy results of Study M05-730 confirmed earlier findings by demonstrating non-inferiority of QD compared to BID LPV/r3; these data also indicated that there were no clinically significant differences in safety or tolerability between treatment groups.
This presentation describes the final 96-week results from Study M05-730.
Objectives
To explore the safety, tolerability and antiviral activity of LPV/r dosed QD or BID in antiretroviral-naïve subjects through 96 weeks of treatment. The emergence of
resistance was also compared in the two treatment groups.
Methods
Study Design
Study M05-730 was an open label, randomized, multicenter, multicountry Phase 3 trial comparing LPV/r QD and BID through 96 weeks of treatment.
664 antiretroviral-naïve, HIV-1-infected subjects with HIV-1 RNA >1,000 copies/mL and any CD4+ T-cell count were randomized 1:1:1:1 to LPV/r QD (SGC),
LPV/r BID (SGC), LPV/r QD (tablet), or LPV/r BID (tablet) for 8 weeks (Figure 1).
At week 8, all subjects receiving SGC were switched to the tablet formulation while maintaining their original dosing schedule.
All subjects also received tenofovir disoproxil fumarate (TDF) 300 mg QD and emtricitabine (FTC) 200 mg QD.
All subjects who were randomized and received at least one dose of study medication were included in the analysis.
For the purposes of the 96-week analysis, subjects were grouped according to LPV/r dosing frequency, regardless of the original formulation administered.
Efficacy Analysis
The proportion of subjects responding in each treatment group was calculated using an intent-to-treat analysis in which noncompleters were considered failures (ITT, NC=F) with a criterion of HIV-1 plasma RNA <50 copies/mL. A non inferiority threshold for the lower margin of the 95% confi dence interval (CI) of
the difference between QD and BID was predetermined to be -12%.
Additional analyses were carried out using an on-treatment approach.
Differences between QD and BID LPV/r treatment groups were tested using Fisher's exact test.
Antiviral activity was also investigated in subgroups of subjects stratifi ed according to baseline disease characteristics.
The mean change in CD4+ T-cell counts from baseline through week 96 was compared between groups using one-way ANOVA.
Safety Analysis
Adverse events were coded according to terminology from the Medical Dictionary for Regulatory Activities (MedDRA®).
The proportion of subjects reporting moderate-to-severe, treatment-related adverse events and the incidence of Grade 3 or greater laboratory abnormalities
through 96 weeks was compared between treatment groups using Fisher's exact test.
The mean changes from baseline through 96 weeks were determined for lipid parameters and compared between groups using one-way ANOVA.
Resistance Analysis
The proportion of subjects with the emergence of resistance mutations to each study drug was evaluated for the LPV/r QD and BID treatment groups.
Subjects had genotype resistance testing performed per protocol if they met the following criteria: HIV-1 RNA > 50 copies/mL after Week 24, and a second
HIV-1 RNA measurement > 400 copies/mL 4 weeks later.
Resistance mutations were defi ned according the IAS-UAS Panel.4 In addition, a more conservative defi nition of lopinavir resistance was used, classifi ed as the
presence of 1 or more of the following protease mutations: I47V/A, G48V, I50V, V82A/F/T/S, I84V, L90M; or the presence of 3 or more of the following protease
mutations: L10F/I/R/V, K20M/R, L24I, V32I, L33F, M36I, M46I/L, F53L, any change to I54, A71V/T, and G73S.
The difference in the incidence of post-baseline resistance was compared between treatment groups using Fisher's exact test.
Figure 1. Study Design and Subject Disposition Through 96 Weeks
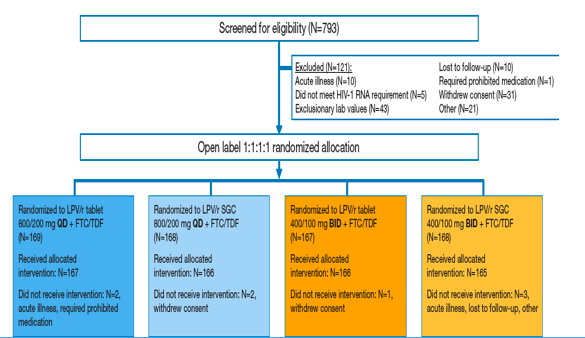
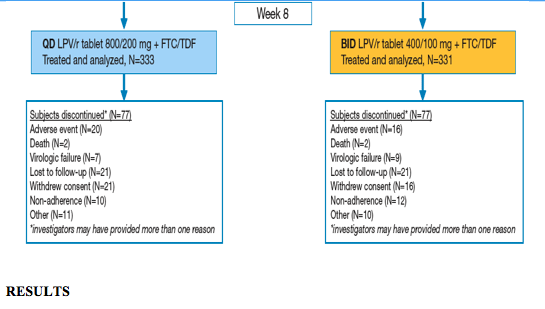
Subject Disposition
Subject disposition through 96 weeks is presented in Figure 1.
Through 96 weeks, 77 subjects from each arm discontinued prematurely, representing 23.1% and 23.3% of QD and BID subjects, respectively.
There were no statistically significant differences between groups in the proportion of subjects discontinuing prematurely for any reason; investigators may have chosen multiple explanations for discontinuation.
- Treatment-emergent adverse events or HIV-related events contributed to discontinuation of 20 QD subjects (6.0%) and 16 BID subjects (4.8%).
- Virologic failure was cited as a reason for discontinuation by the investigator in 7 and 9 subjects from the QD and BID groups (2.1% and 2.7%), respectively.
-- Subject-related factors alone (lost to follow-up, withdrew consent, non-adherence, other) were listed as the reason for discontinuation in 50 QD subjects (15.0%) and 51 BID subjects (15.4%).
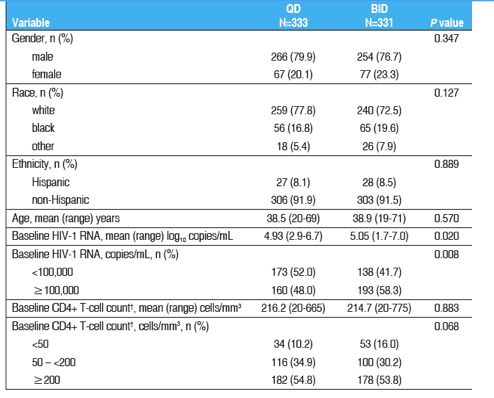
Baseline Demographics
Baseline demographic characteristics are presented in Table 1.
Baseline demographic characteristics were similar between treatment groups.
Subjects in the BID group had a statistically significantly higher baseline mean HIV-1 RNA level, and a statistically greater proportion of BID subjects had baseline HIV-1 RNA ≥100,000 copies/mL.
The mean and distribution of subjects' baseline CD4+ T-cell counts were comparable between groups.
Efficacy
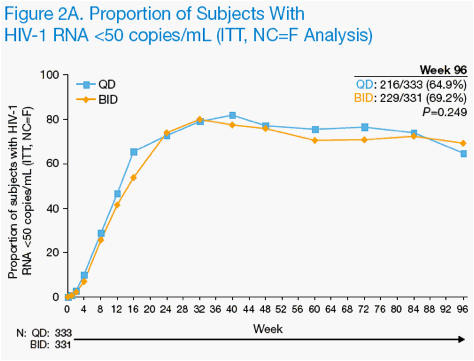
Through 96 weeks of treatment, 216 QD subjects (64.9%) and 229 BID subjects (69.2%, P=0.249) had HIV-1 RNA <50 copies/mL by ITT, NC=F analysis (Figure 2A). The mean difference (95% CI) was -4.3% (-11.5%, 2.8%), demonstrating noninferiority of the QD group compared to the BID group based on the pre-specified noninferiority margin of -12%.
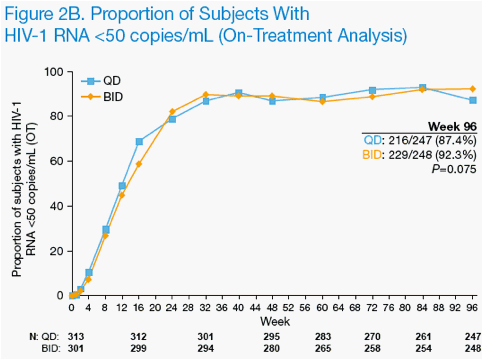
By on-treatment analysis, 216 of 247 QD subjects (87.4%) and 229 of 248 BID subjects (92.3%) exhibited HIV-1 RNA <50 copies/mL (P=0.075) at week 96
(Figure 2B).
Evidence of intermittent low-level viremia, defined as an HIV-1 RNA level ≥50 copies/mL then <50 copies/mL at the subsequent visit, was detected similarly
between treatment groups. There were 91 such instances in 76 QD subjects, while 83 BID-treated subjects experienced these blips on 97 distinct occasions.
Of the individuals exhibiting HIV-1 RNA ≥50 copies/mL at week 96, 16 of 22 QD subjects (72.7%) and 5 of 8 BID subjects (62.5%) had a low level of HIV-1 RNA <200 copies/mL.
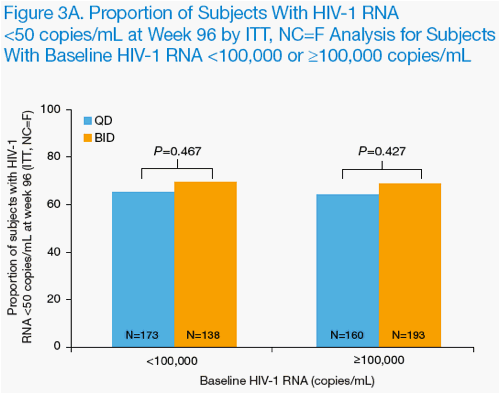
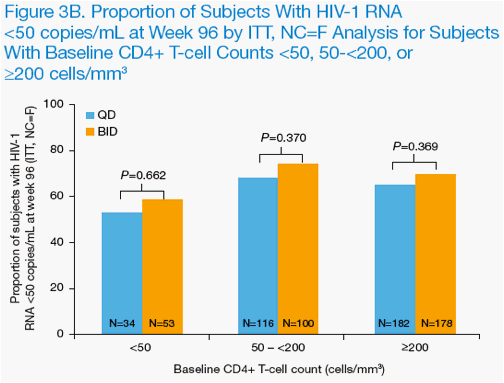
Through 96 weeks of treatment, LPV/r was similarly effective when dosed QD or BID in subjects with high baseline HIV-1 RNA (≥100,000 copies/mL) or low
baseline CD4+ T-cell counts (<50 cells/mm3) by ITT, NC=F analysis (Figure 3A & B).
Similar mean increases in CD4+ T-cell counts from baseline through 96 weeks were observed in QD and BID groups (+238.4 cells/mm3 and +254.0 cells/mm3,
respectively; P=0.269).
Safety
Overall, the incidence of any treatment-emergent adverse event was not statistically significantly different between treatment groups.
Regardless of severity or relationship to study drug, diarrhea occurred with a similar frequency in QD and BID groups.
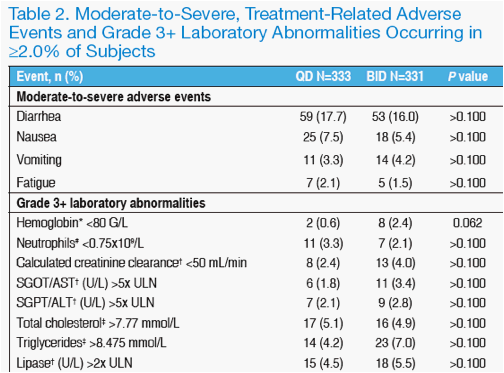
Rates of study drug-related adverse events of moderate or greater severity were comparable between treatment groups (Table 2).
The incidence of Grade 3 or greater laboratory abnormalities was also similar in QD and BID subjects (Table 2).
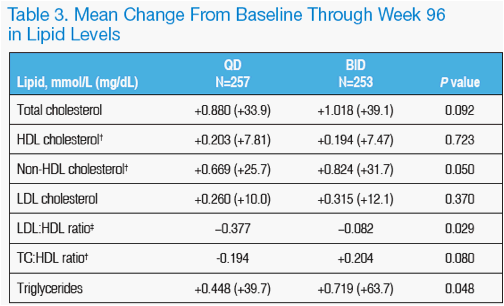
There were statistically significant differences between QD and BID treatment groups in the mean change from baseline through week 96 for triglycerides, non-HDL cholesterol, and the LDL:HDL ratio (Table 3).
The mean change from baseline through week 96 in total cholesterol, LDL cholesterol, HDL cholesterol, or the TC:HDL ratio was not significantly different when LPV/r was administered QD or BID (Table 3).
Resistance
Genotyping data were available for 13 subjects each from the QD and BID treatment groups based on protocol-defined criteria (with the exception of 2/13 QD subjects that experienced viral rebound without a preceding HIV-1 RNA value <50 copies/mL and whose genotype data was provided at the request of the investigator).
The M184V mutation in reverse transcriptase, associated with resistance to FTC and lamivudine, was detected in 4 subjects overall (2 QD, 2 BID).
There was no evidence of mutations proposed to confer resistance to TDF.
Resistance mutations in the protease gene, as defined by the IAS-USA Panel and the more conservative list presented in the Methods, were not detected in any samples.
Reference
1. Molina JM, Podsadecki TJ, Johnson MA, Wilkin A, Domingo P, Myers R, et al. A lopinavir/ritonavir-based once-daily regimen results in better compliance and is non-inferior to a twice-daily regimen through 96 weeks. AIDS Res Hum Retroviruses. 2007;23:1505-14.
2. Eron JJ, Feinberg J, Kessler HA, Horowitz HW, Witt MD, Carpio FF, et al. Once-daily versus twice-daily lopinavir/ritonavir in antiretroviral-naive HIV-positive patients: a 48-week randomized clinical trial. J Infect Dis. 2004;189:265-72.
3. Gathe J, Silva BA, Cohen DE, Loutfy MR, Podzamczer D, Rubio R, et al. A Once-Daily Lopinavir/Ritonavir-Based Regimen Is Noninferior to Twice-Daily Dosing and Results in Similar Safety and Tolerability in Antiretroviral-Naive Subjects Through 48 Weeks. J Acquir Immune Defi c Syndr. 2009;in press.
4. Johnson VA, Brun-Vezinet F, Clotet B, Kuritzkes DR, Pillay D, Schapiro JM, et al. Update of the drug resistance mutations in HIV-1: Fall 2006. Top HIV Med. 2006;14:125-30.
5. King M, Fredrick L, da Silva B, Podsadecki T, Bernstein B. Once-Daily and Twice-Daily Lopinavir/ritonavir-Based Regimens Provide Similar Virologic Response Through 48 Weeks: Results of a Meta-Analysis. 14th Annual BHIVA Conference; 23-25 April; Belfast, Ireland; 2008.
|
| |
|
|
|
|
|