 |
|
|
| |
Safety, Tolerability, Pharmacokinetics and Antiviral Activity of the HCV Polymerase Inhibitor ABT-072 Following Single and Multiple Dosing in Healthy Adult Volunteers and Two Days of Dosing in Treatment-Naïve HCV Genotype 1-Infected Subjects
|
| |
| |
Reported by Jules Levin
HepDART 2009 - 06-10 December 2009 - Kohala Coast, Hawaii, USA
CE Klein,1 D. Cohen,1 RM Menon,1 L Hazan,2 WM Awni,1 YL Chiu,1 EO Dumas,1 LM Larsen,1 TJ Podsadecki,1 A Nada,1 BM Bernstein1
1 Abbott Laboratories, Illinois, USA;
2 Impact Clinical Trials, California, USA
Corresponding author: Rajeev Menon, Abbott, 100 Abbott Park Road, Dept. R4PK, AP13A, Abbott Park, IL 60064, USA
AUTHOR CONCLUSIONS
A dose of ABT-072 160 mg QD for 2 days resulted in a mean decrease of 1.5 log10 HCV RNA.
ABT-072 was safe and well tolerated following single and multiple dosing in healthy subjects and two days of dosing in HCV-infected subjects.
Adverse events were infrequent and generally mild and occurred with similar frequency in subjects receiving ABT-072 or placebo.
The pharmacokinetics of ABT-072 were dose proportional and ABT-072 showed minimal accumulation following multiple dosing.
When ABT-072 was co-administered with ketoconazole, the mean ABT-072 Cmax and AUC values were only 45 and 27% higher as compared to ABT-072 administration without ketoconazole. As the fold increase in AUC was between 1.25 and 2, based on FDA guidance, ABT-072 is not a sensitive substrate of CYP3A.
These findings support continued development of ABT-072 as a potential once daily non-nucleoside polymerase inhibitor.
Future clinical studies will be conducted with a solid tablet formulation that supports escalation and evaluation of higher doses.
Background
ABT-072 is a sulfonamide palm polymerase inhibitor of the hepatitis C virus (HCV) being developed for the treatment of HCV genotype 1 infection.
ABT-072 has shown inhibitory concentrations in the nanomolar range in genotypes 1a and 1b subgenomic replicon systems.
Three studies evaluated the safety, tolerability and pharmacokinetics of
oral doses of ABT-072:
- Study 1 was a first-in-human study which evaluated ABT-072 single ascending doses (SAD) in healthy subjects.
- Study 2 evaluated multiple doses in HCV genotype 1-infected subjects.
- Study 3 evaluated multiple doses of ABT-072 alone and ABT-072 coadministered with ketoconazole in healthy subjects.
OBJECTIVES
Study 1: Assess the safety, tolerability, and pharmacokinetics of escalating, single, oral doses of ABT-072 under nonfasting conditions in healthy adult subjects.
Study 2: Assess the safety, tolerability, antiviral activity, and pharmacokinetics of oral doses of ABT-072 under nonfasting conditions over two days in HCV genotype 1-infected adult subjects.
Study 3:
- Primary: Determine multiple-dose safety, tolerability and pharmacokinetics of ABT-072 under nonfasting conditions in healthy adult subjects.
- Secondary: Determine the effect of single-dose administration of
ketoconazole on steady state ABT-072 pharmacokinetics.
METHODS
Study Designs
The protocols were reviewed and approved by an IRB prior to the start of the study. All subjects who participated in the studies gave written informed consent prior to starting any study procedures.
Safety, tolerability, and preliminary pharmacokinetic results were assessed in each group prior to the start of the next group.
Study 1 (Healthy Subjects)
Study 1 was a first-in-human, single-ascending dose (SAD), blinded, placebo-controlled, nonfasting study conducted according to a randomized, sequential design.
The study was conducted in 10 sequential groups in healthy adult male and female subjects. Approximately 10 subjects in each group were randomized in a 4:1 fashion (ABT-072:placebo) to receive a single dose following a moderate-fat meal.
Doses were administered in the form of capsules or as the capsule contents suspended in water or a lipid vehicle. Doses from 10 to 80 mg were initially dosed using a capsule. Due to less-than-expected exposures from the intact capsule dosed orally, dosing was continued by suspending the capsule contents in a lipid vehicle. Included in this poster are pharmacokinetic results from dosing ABT-072 in the lipid vehicle (80 mg, 160 mg and 320 mg) and safety from all subjects dosed with ABT-072 (as a capsule, capsule contents suspended in water and capsule contents suspended in lipid).
Study 2 (HCV-Infected Subjects)
Adult male and female subjects (N=6) infected with HCV genotype 1 were randomized in a blinded 2:1 fashion (ABT-072:placebo) to receive two, once-daily (QD) doses of ABT-072 160 mg following a moderate-fat meal.
All subjects were offered post-study treatment with pegylated interferon and ribavirin for up to 48 weeks.
Study 3 (Healthy Subjects)
Study 3 was a Phase 1, multiple-dose, blinded, placebo-controlled, nonfasting study conducted in healthy adult male and female subjects(N=32) according to a randomized, sequential design.
In Group 1 (80 mg), 12 subjects were randomized in a 2:1 ratio (ABT-072:placebo). In Groups 2 (160 mg) and 3 (320 mg ABT-072) 10 subjects per group were randomly assigned to receive ABT-072 or matching placebo in a 4:1 ratio.
ABT-072 was dosed QD in Group 2 and 3 for 10 days and in Group 1 for 11 days. For Group 1, on Day 11, ketoconazole 400 mg was dosed 30 minutes prior to administration of ABT-072 80 mg.
Pharmacokinetic Assessments: Studies 1, 2 and 3
In Studies 1 and 2, complete pharmacokinetic profiles for ABT-072 were drawn up to 72 hours post last dose. In Study 3 complete pharmacokinetic profiles for ABT-072 were drawn on Days 1 and 10 (and Day 11 for Group 1) and trough samples were collected immediately prior to the morning dose on Days 2, 3, 5, 7, 8, and 9.
ABT-072 concentrations were determined using a liquid chromatography tandem mass spectroscopic method (LC-MS/MS) with a lower limit of quantitation (LLOQ) of 0.325 ng/mL.
Pharmacokinetic and statistical analysis was conducted using SAS version 9.13.
The effect of ketoconazole on the steady state ABT-072 pharmacokinetics was assessed by comparing log-transformed Cmax and AUC24 on Day 11 (with ketoconazole) to those on Day 10 (without ketoconazole) using analysis of variance (ANOVA). Least square mean ratios and 90% confidence intervals were calculated to quantify the effect of ketoconazole on ABT-072.
Efficacy Assessments: Study 2
Serum HCV levels were determined by the central laboratory using Roche COBAS Taqman® RT-PCR assay (LOQ= 25 IU/mL, LOD = 10 IU/mL).
Safety and Tolerability Assessments: Studies 1, 2 and 3
Safety and tolerability assessments included physical examination, vital sign measurements and clinical laboratory tests.
12-lead Electrocardiogram (ECG) were recorded in triplicate at multiple time-points before and after dosing. Twelve lead continuous cardiac monitoring was also conducted on all subjects in Study 1.
Adverse events were monitored throughout the study.
Adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA 11.1).
RESULTS
Table 1. Demographic Summary for all Subjects
Dosed with ABT-072/Placebo in Lipid Vehicle
SD = Standard Deviation.
In addition to the subjects included in this table, 64 healthy subjects were dosed in Study 1 using ABT-072 in a capsule or capsule contents suspended in water at doses of 10 to 80 mg.
Pharmacokinetic Results: Study 1
(Single-Dose, Healthy)
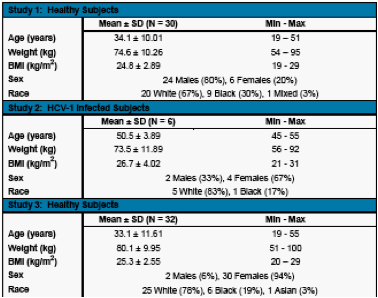
The mean concentration time profiles and the mean pharmacokinetic parameters for ABT-072 in healthy subjects following a single dose of ABT-072 are shown in Figure 1 and Table 2, respectively.
Single dose administration of ABT-072 in a lipid vehicle resulted in dose proportional increases in exposure from 80 to 320 mg.
Maximum concentrations were achieved within 5-9 hours of dosing ABT-072 in a lipid vehicle and the mean terminal half-life was 7-9 hours.
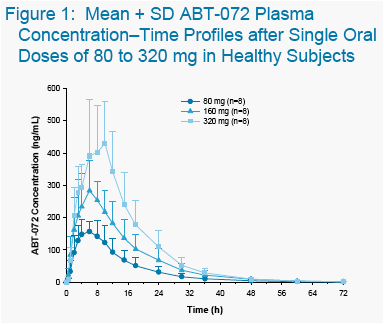
Table 2. Mean ± SD Pharmacokinetic Parameters of
Single Doses of ABT-072 to Healthy Subjects and
2-Day Doses to HCV+ Subjects
Values for Cmax, Tmax, AUC24, dose-normalized Cmax and dose-normalized AUC24 were calculated after Day 1 dosing and values for t1/2 were calculated after Day 2 dosing.
Presented as harmonic mean and pseudo SD.
AUCτ is presented for Study 1; AUC24 is presented for Study 2.
Pharmacokinetic Results: Study 2
(HCV-infected Subjects)
ABT-072 was dosed as 160 mg (n=4) or placebo (n=2) once daily for 2 days in HCV genotype 1-infected subjects.
Pharmacokinetic parameters of ABT-072 following 2 days of 160 mg QD administered in a lipid vehicle in Study 1 to HCV-infected subjects are shown in
Table 2.
The mean Cmax, AUC and t1/2 values in HCV+ patients were similar to those in healthy subjects.
Efficacy Results: Study 2
(HCV-infected Subjects)
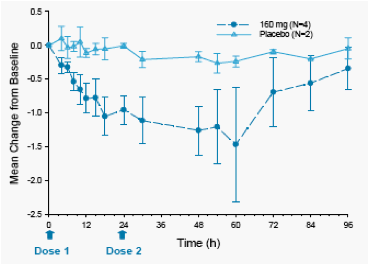
Figure 2 displays the mean change from baseline in log10 HCV RNA following 2-day monotherapy in HCV-infected subjects.
Mean maximum HCV RNA change from baseline for ABT-072 160 mg QD for 2 days was -1.5 log10 compared to -0.3 log10 for placebo.
Three genotype 1a subjects and one genotype 1b subject were dosed with ABT-072. The 3 genotype 1a subjects had a mean maximal decrease in viral load of 1.19 log; the one genotype 1b subject had a maximum decrease in viral load of 2.3 log.
Figure 2. Mean ± SE Change from Baseline in Log10
HCV RNA
Pharmacokinetic Results: Study 3
(Multiple-Dose, Healthy)
The mean concentration time profiles and the mean pharmacokinetic parameters for ABT-072 in healthy subjects following 10-day QD dosing of ABT-072 are shown in Figure 3 and Table 4, respectively.
An approximate dose-proportional increase in Cmax and AUC24 was observed following multiple ABT-072 dosing of 80, 160, and 320 mg QD for 10 days.
Mean Cmax and AUC24 values on Day 10 were 7~30% higher compared to Day 1 following multiple ABT-072 dosing of 80 to 320 mg QD for 10 days, suggesting minimal accumulation.
Consistent with minimal accumulation, steady state levels of ABT-072 were reached by Day 2 following ABT-072 80 to 320 mg QD dosing.
Figure 3: Mean ± SD ABT-072 Plasma Concentration-Time Profiles Following ABT-072 QD Dosing for 10 Days
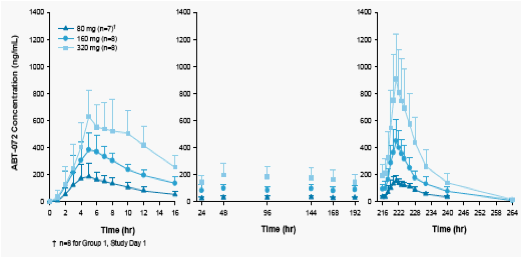
Table 4. Mean ± SD Pharmacokinetic Parameters of ABT-072 Following 10-day Dosing in Healthy Subjects
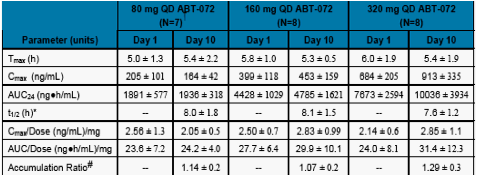
N=8 for Group 1 on Study Day 1.
Harmonic Mean and Pseudo %CV
Calculated as a Ratio of AUC24 Day 10/Day 1
N=8 for Group 1 on Study Day 1.
Harmonic Mean and Pseudo %CV
N=8 for Group 1 on Study Day 1.
Calculated as a Ratio of AUC24 Day 10/Day 1
Effect of Ketoconazole Dose on ABT-072 Pharmacokinetics
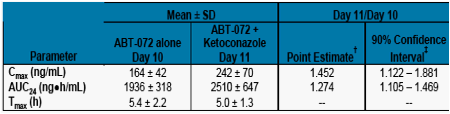
The mean concentration time profiles and the mean pharmacokinetic parameters for ABT-072 dosed with and without ketoconazole are shown in Figure 4 and Table 5, respectively.
Following coadministration of ABT-072 80 mg with ketoconazole, a strong CYP3A inhibitor,
The mean ABT-072 Cmax and AUC24 values were 45 and 27% higher compared to ABT-072 administered without ketoconazole.
Based on FDA guidance, ABT-072 is not considered a "sensitive substrate" of CYP3A as the fold increase in AUC was between 1.25 and 2.
Figure 4. Mean + SD Concentration Time Profiles for ABT-072 80 mg Administered Alone and with Ketoconazole
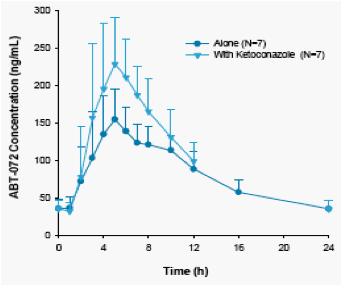
Table 5. Mean ± SD Pharmacokinetic Parameters of
ABT-072 with and without Ketoconazole
Safety Results
ABT-072 was well tolerated with few adverse events (AEs).
There were no severe or serious AEs and no premature discontinuations due to AEs in these studies.
The most common drug-related AEs in the 101 healthy subjects who received ABT-072 in the single and multiple dose studies were headache (5%), diarrhea (5%), abdominal pain (3%), and fatigue (3%).
Of the 101 healthy subjects who received ABT-072, 48 received ABT-072 in a lipid vehicle. Among these subjects, diarrhea, fatigue and headache were each
reported by four subjects (4/48, 8.3%) and abdominal pain was reported by 3 subjects (3/48, 6.3%).
No apparent dose effect was observed in the frequency or character of adverse events
No clinically significant laboratory abnormalities were observed.
ABT-072 was similarly safe and well tolerated in the 4 HCV genotype 1-infected subjects. Headache was the only AE reported by more than 1 subject (n=2).
Pharmacokinetic Results
Multiple-Dose Pharmacokinetics of ABT-450 Co-administered with Ritonavir
Mean and SD plasma concentration-time profiles and pharmacokinetic parameters for ABT-450 after administration of multiple oral doses of ABT-450/r are presented in Figure 1 and Table 2.
The mean ABT-450 Cmax and AUC values increased in a nonlinear fashion following multiple dosing.
The mean ABT-450 trough concentrations showed an initial increase followed by a gradual decrease which stabilized around Day 9 through Day 12. The initial increase in trough concentrations was higher for the BID dosing regimen than that for the QD dosing regimen.
The ABT-450 exposures on Day 14 were approximately 2- to 3-fold higher than that on Day 1 following multiple dosing of ABT-450/r.
The terminal half-life values following multiple dosing were similar to that observed following single dosing,1 approximately 4-7 hours.
Figure 1. Mean + SD Plasma Concentration-Time Profiles and Trough Concentrations for ABT-450 Following ABT-450/r QD and BID Dosing for 14 Days>
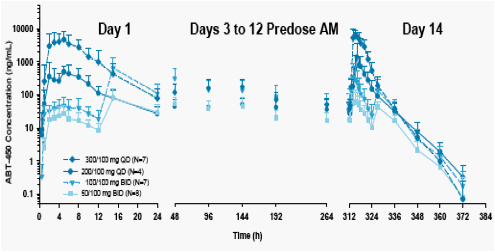
Table 2. Mean ± SD ABT-450 Pharmacokinetic Parameters on Day 1 and Day 14 After Administration of Multiple Doses of ABT-450/r
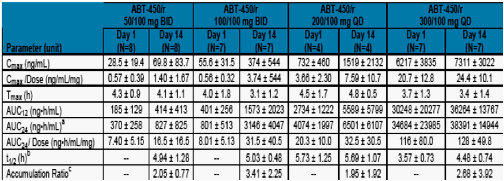
a. For bid dosing regimen the AUC24 was calculated as 2*AUC12.
b. Presented as the harmonic mean and pseudo standard deviation.
c. Ratio of AUCτ from Day 14 to Day 1.
Multiple Dose Pharmacokinetics of Ritonavir Co-administered with ABT-450
Mean and SD plasma concentration-time profiles and pharmacokinetic parameters for ritonavir after administration of multiple oral doses of ABT-450/r are presented in Figure 2 and Table 3.
Figure 2. Mean and SD Ritonavir Plasma Concentration-time Profiles Following Multiple Oral Doses of ABT-450/r for 14 Days
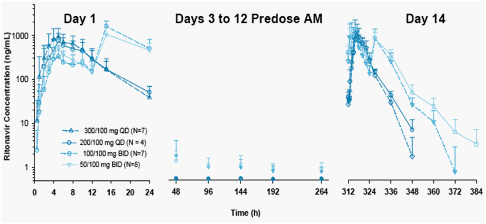
Table 3. Mean + SD Ritonavir Pharmacokinetic Parameters on Day 1 and Day 14 After Administration of Multiple Doses of ABT-450/r
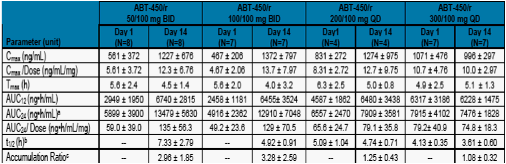
a. For bid dosing regimen the AUC24 was calculated as 2*AUC12.
b. Presented as the harmonic mean and pseudo standard deviation.
c. Ratio of AUCτ from Day 14 to Day 1.
Safety Results
ABT-450/r dosed QD or BID for 14 days in healthy volunteers was generally well tolerated.
Adverse events were mostly mild, and no serious or severe adverse events were reported.
There were no clinically significant dose-related changes from baseline in vital signs or ECG parameters.
Adverse Events
20/30 healthy volunteers receiving ABT-450 experienced study drug-related
adverse events during the 14-day dosing period. The most common adverse events reported in ≥3 subjects were fatigue (n=8), elevated bilirubin (n=8), headache (n=4), and diarrhea (n=3). All events resolved by the end of the study follow-up period.
3 subjects were prematurely discontinued from study due to adverse events:
- 2 subjects were discontinued due to increased ALT values that were above the normal range, but did not meet the criteria for potentially clinically significant (All values were <3 x upper limit of normal). These ALT increases were not associated with concomitant increases in bilirubin levels.
-- 1 subject was discontinued due to mild indirect bilirubin elevations: both parameters returned to baseline levels within a few days. These elevations were not associated with jaundice and other liver test abnormalities.
Laboratory Abnormalities
Laboratory abnormalities were mostly Grade 1 in severity.
Asymptomatic, transient increases in indirect bilirubin without concomitant abnormalities in direct bilirubin or other liver function tests were observed.
Maximum indirect bilirubin value: 2.7 mg/dL
Bilirubin levels normalized in all subjects with ongoing dosing. Figure 3 shows the indirect bilirubin profiles for all subjects at the highest dose in the study indicating that indirect bilirubin levels came back to baseline with continued dosing in all subjects.
Indirect bilirubin levels correlated strongly with measured ABT-450 trough concentration over time, whereas they did not correlate with Day 1 or Day 14 Cmax or AUC.
The increase in indirect bilirubin was described by a sigmoidal Emax model. Figure 4 shows the relationship between indirect bilirubin and ABT-450 Ctrough. Table 4 shows the population parameters and confidence intervals for the Emax model.
Figure 3. Indirect Bilirubin-Time Profiles for all Subjects at the Highest Dose of ABT-450/r (300/100 mg) in the Study following 14 Days of Daily Dosing
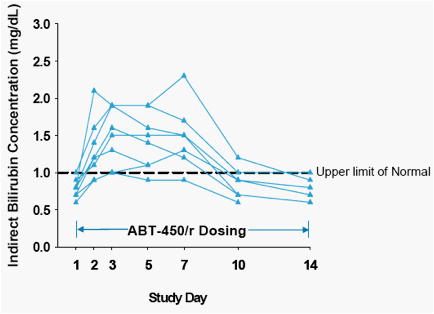
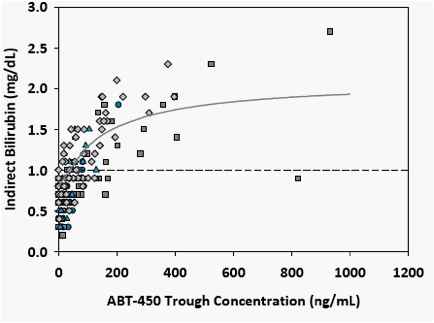
Figure 4. Relationship Between Indirect Bilirubin Levels and ABT-450 Trough Concentrations
Reference
1. Menon R, et al. Pharmacokinetics and Tolerability of the HCV Protease Inhibitor ABT-450 Following Single Ascending Doses in Healthy Adult Volunteers with and Without Ritonavir. Annual Meeting of HepDART 2009; Poster 57.
|
| |
|
|
|
|
|