| |
Antiviral activity and safety of LB80380 in hepatitis B e antigen-positive chronic hepatitis B patients with lamivudine-resistant disease
|
| |
| |
Hepatology Jan 2010
"In conclusion, the results of the present study suggest that LB80380 at doses of up to 240 mg is safe, well tolerated, and effective at reducing viral load in adult patients with lamivudine-resistant chronic hepatitis B for a period of 12 weeks. LB80380 showed promising results causing profound viral suppression after only 12 weeks of therapy. Further clinical investigation focusing on long-term treatment should be performed to verify the effects and safety of this compound for the treatment of chronic hepatitis B viral infection in both treatment-naïve and lamivudine-resistant patients.......The mean reduction from baseline viral load was as follows: group 1 = 2.81 log10 copies/mL (95% CI 2.17-3.45); group 2 = 3.21 log10 copies/mL (95% CI 2.50-3.93); group 3 = 3.92 log10 copies/mL (95% CI 3.36-4.49); group 4 = 4.16 log10 copies/mL (95% CI 3.51-4.81); and group 5 = 4.00 log10 copies/mL (95% CI 3.79-4.21)."
Man-Fung Yuen 1, Kwang-Hyub Han 2, Soon-Ho Um 3, Seung Kew Yoon 4, Hye-Ryon Kim 5, John Kim 5, Chung Ryeol Kim 5, Ching-Lung Lai 1
1Department of Medicine, Queen Mary Hospital, The University of Hong Kong, Hong Kong
2Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Korea
3Department of Internal Medicine, Korea University College of Medicine, Seoul, Korea
4Department of Internal Medicine, College of Medicine, The Catholic University of Korea, Seoul, Korea
5LG Life Sciences, Seoul, Korea
email: Ching-Lung Lai (hrmelcl@hkucc.hku.hk)
*Correspondence to Ching-Lung Lai, Department of Medicine, Queen Mary Hospital, The University of Hong Kong, Pokfulam Road, Hong Kong
This study has been registered (registration number NCT00895596) in ClinicalTrials.gov (URL http://clinicaltrials.gov/ct2/show/NCT00895596?term=LB80380&rank=1).
This study was supported by LG Life Sciences which also provided the drug LB80380. The data collection and data report were carried out by an independent data management company, Covance Pty Limited, Australia. The analysis and the writing of the manuscript were mainly performed by the first two authors as well as the corresponding author. The drug company employees read and commented on the manuscript, and gave their approval.
Potential conflicts of interest: M. F. Y. and C. L. L. have received speakers' honoraria from LG Life Sciences. H. R. K. and C. R. K. are full-time employees of LG Life Sciences. J. K. is a former employee of LG Life Sciences.
These authors contributed equally to this work.
||fax: (852)-28162863.
Funded by:
LG Life Sciences
Korean Ministry for Health, Welfare, and Family Affairs
Anadys Pharmaceuticals, Inc.
Abstract
We aimed to determine the antiviral activity and safety of a new nucleotide analogue, LB80380, in chronic hepatitis B (CHB) patients with lamivudine-resistant virus. Sixty-five patients with lamivudine-resistant virus were randomized to receive five ascending daily doses (30, 60, 90, 150, 240 mg) of LB80380. LB80380 was given together with lamivudine for the first 4 weeks, followed by 8 weeks of LB80380 monotherapy. This was then followed by 24 weeks of adefovir. Hepatitis B virus (HBV) DNA levels, serology, liver biochemistry, and safety were monitored. The extent of the HBV DNA reduction at week 12 was dose-dependent.
The mean reduction from baseline was 2.81, 3.21, 3.92, 4.16, and 4.00 log10 copies/mL for the five ascending dose groups.
The dose-proportionate effect was statistically significant (P < 0.001) with a decrease of HBV DNA levels by an average of 1.54 log10 copies/mL for every 1-unit increase in log10 dose of LB80380. In 93.4% of patients, HBV DNA decreased by >2 log10 copies/mL, and 11.5% of patients had undetectable HBV DNA levels (<300 copies/mL) by week 12. HBV DNA suppression was maintained during the 24 weeks of adefovir treatment. Hepatitis B e antigen seroconversion and normalization of alanine aminotransferase were seen in 14.6% and 24.6% of patients, respectively, at week 12; 44.6% of patients experienced mild and self-limiting adverse events, none of which were attributed to the study drug.
Conclusion: LB80380 at doses of up to 240 mg is safe, well tolerated, and effective at reducing viral load in CHB patients with lamivudine-resistant virus for a period of 12 weeks.
Article Text
Treatment for chronic hepatitis B (CHB) disease is rapidly evolving after the introduction of nucleoside/nucleotide analogs (NAs). The first nucleoside analog lamivudine (L-nucleoside) was licenced in 1998,[1] and four more NAs were approved subsequently: adefovir in 2002, entecavir in 2005, telbivudine in 2006, and, most recently, tenofovir in 2008.[2] Apart from having a wider option of drug treatment, the treatment strategy has also been conceptually changed, aiming at prolonged viral suppression to achieve reduction in the rate of development of cirrhosis and hepatocellular carcinoma.[3]
The main limitation of using NAs is the emergence of drug resistance. Because lamivudine was the first available NA, it has been used extensively worldwide. It has been shown that the chance of lamivudine resistance is approximately 76% after 5 years of treatment.[4] Adefovir resistance also occurs in 20%-29% of treatment-naïve patients after 5 years of therapy. Although adefovir is effective in suppressing lamivudine-resistant hepatitis B virus (HBV), its potency is only modest at the licensed dosage. Adefovir treatment for 48 weeks for patients with lamivudine-resistant HBV is associated with a 4-log HBV DNA level reduction and 53% chance of normalization of alanine aminotransferase (ALT) levels.[5] Lamivudine-resistant HBV is partially refractory to entecavir treatment requiring increased doses of entecavir. In addition, entecavir resistance occurs in 51% of patients with lamivudine-resistant HBV after 5 years of treatment.[6] Tenofovir is highly effective for treatment-naïve patients.[7] Tenofovir is also effective in patients with lamivudine-resistant virus, although more long-term data on the development of tenofovir resistance is still not available.
It has been shown in in vitro studies and case reports that different NAs are effective for drug-resistant HBV to other NAs. For instance, tenofovir is effective against adefovir-resistant strains[8] and adefovir is effective against entecavir resistant strains.[9] There is therefore a need to develop newer NAs to provide complementary and possibly better viral suppression for both treatment-naïve patients and patients with drug-resistant HBV.
LB80380 is a new acyclic nucleotide phosphonate with chemical similarity to adefovir and tenofovir. It is the prodrug of LB80331, which in turn will be metabolized to LB80317, the active metabolite with antiviral effect for HBV after further intracellular phosphorylation to the triphosphate form.[10] Experiments conducted in Huh7 cells show that there is no reduction in mitochondrial DNA and no lactic acid accumulation with LB80331.[11]
According to a phase Ib dose escalation study in treatment-naïve patients, 4-week treatment of LB80380 is associated with a 3- to 4.2-log reduction of HBV DNA and the dose ranged from 30 to 240 mg daily.[12] In an in vitro experiment, this drug has also been shown to be effective against HBV mutants resistant to lamivudine, adefovir, entecavir, and telbivudine.[13]
We report the results of an open-label, multicenter dose escalation study to assess the antiviral activity and safety of LB80380 for 12 weeks in CHB patients with lamivudine-resistant disease.
Results
Baseline Characteristics.
The baseline demographics, liver biochemistry, HBV DNA levels, and pattern of lamivudine resistance for the PP population are presented in Table 1.
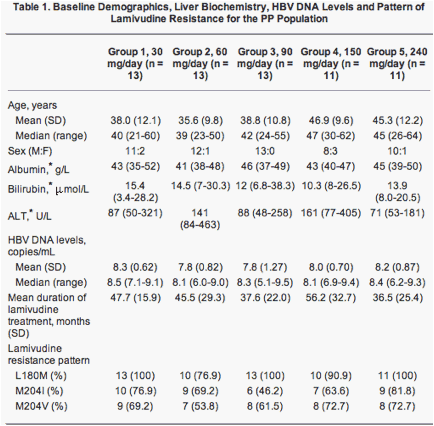
Effect of LB80380 on Serum HBV DNA.
At week 12, there was a decrease from baseline in serum HBV DNA levels in all dose groups in the PP population (Table 2). The mean serum HBV DNA at week 12 for the five dose groups was: group 1 = 5.50 log10 copies/mL (standard deviation [SD] 1.34); group 2 = 4.60 log10 copies/mL (SD 1.24); group 3 = 3.86 log10 copies/mL (SD 1.14); group 4 = 3.84 log10 copies/mL (SD 1.33); and group 5 = 4.19 log10 copies/mL (SD 0.99). The extent of the reduction in viral load from baseline to week 12 was dose-dependent up to the 150 mg daily dose (Table 2). The mean reduction from baseline viral load was as follows: group 1 = 2.81 log10 copies/mL (95% CI 2.17-3.45); group 2 = 3.21 log10 copies/mL (95% CI 2.50-3.93); group 3 = 3.92 log10 copies/mL (95% CI 3.36-4.49); group 4 = 4.16 log10 copies/mL (95% CI 3.51-4.81); and group 5 = 4.00 log10 copies/mL (95% CI 3.79-4.21).
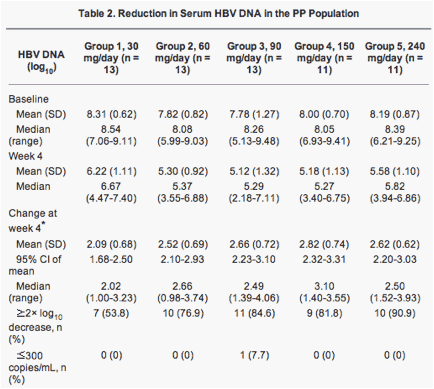
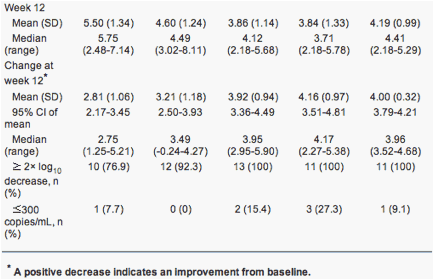
The reduction of HBV DNA levels of the five groups of the PP population over the whole study period are illustrated in Fig. 2. In all dose groups in the PP population, mean HBV DNA copy number decreased incrementally from baseline to week 12. In group 1, patients achieved a peak reduction in mean HBV DNA at week 24, after which mean HBV DNA levels remained relatively constant. In group 2, patients achieved a peak reduction at around week 16, after which mean HBV DNA decreased slightly. In group 3, the reduction in mean HBV DNA was most significant between baseline and week 12, yet HBV DNA copy number continued to decrease slightly up to week 36. In group 4, patients achieved a peak reduction in mean HBV DNA at week 12, after which time it remained relatively constant. In group 5, patients achieved a peak reduction in mean HBV DNA at week 12, the mean HBV DNA copy number increasing thereafter from the week 12 level between weeks 16 and 36.
Increasing doses of LB80380 produced greater mean reductions in HBV DNA levels as illustrated in Fig. 3. The dose proportionality constant was 1.54 (95% CI 0.75-2.33). As such, HBV DNA levels would have a decrease by an average of 1.54 log10 copies/mL for every 1 unit increase in log10 dose of LB80380. The dose-proportionate effect of LB80380 on HBV DNA was statistically significant (P < 0.001).
All except four patients (57/61 [93.4%]) had a decrease from baseline to week 12 in serum HBV DNA copy number of 2 log10 units or more. At week 12, 11.5% of the PP population (7/61) had undetectable HBV DNA levels (1 in group 1; 2 in group 3; 3 in group 4; 1 in group 5).
The HBV DNA levels and reduction of HBV DNA levels from baseline at week 4 are also described in Table 2. The dose proportionality constant was 0.65, with 95% CI 0.09-1.22 (Fig. 3). HBV DNA levels would have a decrease by an average of 0.65 log10 copies/mL for every 1 unit increase in log10 dose of LB80380. The dose-proportionate effect of LB80380 on HBV DNA was statistically significant (P = 0.025).
In all except group 5, the highest-dose group, the antiviral effect of 12 weeks of treatment with LB80380 was maintained during the 24-week follow-up period while the patients were maintained on adefovir. In groups 1, 2, and 3, there was a slight reduction in mean HBV DNA level between week 12 and week 24: group 1 = 1.40 log10 copies/mL (SD 1.08); group 2 = 0.81 log10 copies/mL (SD 0.82); and group 3 = 0.32 log10 copies/mL (SD 0.40). In groups 4 and 5 there was a slight increase in mean HBV DNA level: group 4 = -0.06 log10 copies/mL (SD 0.55) and group 5 = -0.64 (SD 0.85).
Thirteen patients experienced a virologic rebound during the whole study period. All the episodes of rebound occurred after switching to adefovir. Of these 13 patients, six (one from group 3, five from group 5) had virologic rebound 4 weeks after switching from LB80380 to adefovir. The remaining seven patients (three from group 2, one from each of the other groups) had the virologic rebounds at variable time points during the 24 weeks of adefovir treatment.
HBeAg Seroconversion and Loss of HBsAg.
Excluding patients from group 1 in whom serology testing was not conducted at week 12 before protocol amendment, seven patients in the PP population (7/48 [14.6%]) achieved HBeAg seroconversion at week 12 (one in group 2, three in group 3, two in group 4, and one in group 5). No dose-dependent effect of LB80380 on HBeAg seroconversion was observed (P = 0.85). None of the study patients lost HBsAg at week 12.
ALT Normalization.
At week 12, 24.6% (15/61) of patients in the PP population showed normalization of ALT (three in group 1, one in group 2, five in group 3, five in group 4, one in group 5). No dose-dependent effect of LB80380 on ALT normalization was observed (P = 0.90).
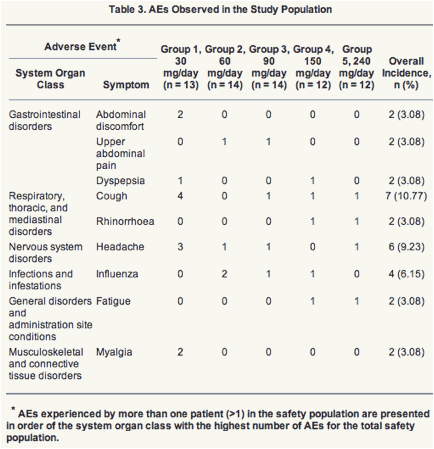
Adverse Events.
Twenty-nine out of 65 (44.6%) patients experienced a total of 65 adverse events during the period of observation. Most of these events appeared to occur in group 1, where 69.2% (9/13) of the patients experienced at least one AE. None of the 65 events were considered to be related to study medication. The most frequently occurring AEs are listed in Table 3. There were no serious or life-threatening (grade 4) AEs. There were no withdrawals due to an AE. The majority (56/65 [86.2%]) of the AEs were of mild (grade 1) intensity. There were two AEs of severe (grade 3) intensity.
Eighteen patients had increases in ALT levels during the entire study period (four in group 1, three in group 2, three in group 3, five in group 4, and three in group 5). One group 3 patient exhibited hepatic flare following the end of treatment with lamivudine, with ALT levels increasing from 298 U/L (5.6 x ULN) at week 4 to 584 U/L (11.0 x ULN) at week 8. This patient already had very high ALT values of 263 U/L at screening and 258 U/L at baseline. This patient's ALT level decreased to 73 IU/L at week 12 and normalized by the end of the study.
Mean change in estimated CrCl from baseline was variable, within dose groups as well as between dose groups at week 12 (end of LB80380 treatment). The mean changes of CrCl from baseline to week 12 for groups 1 to 5 were -5.67 (SD 9.58), 0.52 (SD 9.14), 1.75 (SD 12.0), 4.87 (SD 10.65), and 1.99 (SD 12.26) mL/minute, respectively. The mean CrCl at baseline and week 12 were 102.36 mL/minute (SD 24.96) and 96.68 mL/minute (SD 22.14) for group 1; 94.65 mL/minute (SD 13.05) and 95.18 mL/minute (SD 14.44) for group 2; 96.46 mL/minute (SD 29.33) and 98.21 mL/minute (SD 25.86) for group 3; 87.35 mL/minute (SD 20.27) and 92.23 mL/minute (SD 24.79) for group 4; and 94.86 mL/minute (SD 21.23) and 96.85 mL/minute (29.67) for group 5, respectively. There were no significant differences in the CrCl between the values at baseline and week 12 in all the five groups (P > 0.05). The exact CrCl values at baseline, week 12 (end of LB80380 treatment), and week 36 (end of adefovir treatment) for all individual patients in the five groups are depicted in Fig. 4. Two patients in group 1 experienced an increase in creatinine greater than the predetermined amount at week 28 and week 36, respectively. The CrCl were 78.6 mL/minute and 101.1 mL/minute, respectively.
Discussion
According to our previous study of LB80380 given for 4 weeks in treatment-naïve CHB patients, there is a dose-proportional effect on HBV DNA reduction with an increasing dose.[12] The maximal HBV DNA suppression with 4 logs HBV DNA reduction after 4 weeks is achieved with the dose of equal or higher than 60 mg daily. In the current study, for lamivudine-resistant disease, a dose-proportional effect was also demonstrated with increasing doses of LB80380 up to 150 mg daily. This could be mathematically expressed by the dose-proportional constants for every single log unit increase in the dose for week 4 and 12 (Fig. 3). The maximal mean HBV DNA reduction was achieved at the dose of 150 mg daily (group 4) (Table 2, Fig. 2), with 4.16 logs copies/mL reduction after only 12 weeks of treatment. The mean HBV DNA suppression after 1-year treatment of adefovir and of entecavir (1 mg daily) in lamivudine-resistant patients are 4.0 logs copies/mL and 5.1 logs copies/mL, respectively.[5][14] This suggests that greater viral suppression may be achieved by LB80380. In the present study, there was an increase of median HBV DNA at 16 weeks (i.e., 4 weeks after switching from LB80380 to adefovir) in group 5 (Fig. 2). All 13 episodes of virologic rebound occurred after switching to adefovir. The highest dose of LB80380 (group 5) had earlier virologic rebound. This was presumably related to the greater suppression of HBV DNA with this dosage. However, it should be noted that the study was not empowered statistically to compare the efficacy between these two antiviral agents. The HBV DNA reduction achieved by LB80380 and tenofovir appears to be comparable. The mean HBV DNA reduction at week 12 was 4.16 logs copies/mL for LB80380 and 4.5 logs copies/mL for tenofovir.[15] However, head-to-head comparative studies must be performed for more definite conclusions.
It has been shown in in vitro studies that, of nine mutants resistant to lamivudine, adefovir, entecavir, or telbivudine tested, LB80380 is as potent against six of these as the wild-type virus.[13] Two other mutants have a small decrease (<7-fold) in sensitivity to LB80380. Thus, LB80380 may have an important role in controlling single drug-resistant HBV. Though adefovir and tenofovir are both acyclic phosphonates, a preliminary study has shown that tenofovir is effective in suppressing the HBV DNA levels in adefovir-resistant HBV.[7] It is therefore worthwhile to further investigate the efficacy of LB80380 for HBV resistant to either nucleoside analogues or nucleotide analogues. This can provide more complementary coverage of drug-resistant HBV.
We did not observe a dose-proportional effect on HBeAg seroconversion, loss of HBsAg, and ALT normalization. This is likely related to the statistical limitations because of the limited number of patients achieving these events within a short period of treatment. Concerning the ALT normalization, we cannot exclude the possibility of drug-induced ALT elevation even though there is no documented liver derangement associated with LB80380 in preclinical or phase I studies.
LB80380 was safe for the study period of 12 weeks. All the adverse events were considered to be not related to the medication. The most frequently reported adverse events were cough and headache and occurred primarily in the lowest-dose group (group 1), suggesting no evidence for a dose-related trend.
Comparing baseline CrCl with posttreatment CrCl, there is no clear finding to conclude that LB80380 treatment has a negative impact on renal function in terms of CrCl. Some patients' CrCl decreased after initiation of LB80380 treatment (data not shown); however, the number of patients with decreased CrCl was not increased dose-dependently, and most of the changes were considered as normal fluctuation.
The present study had two limitations. First, it did not recruit a group of patients with lamivudine-resistant disease continuing with lamivudine monotherapy to act as a control population. Second, the duration of LB80380 treatment was only 12 weeks. A longer study would allow better surveillance for possible viral mutations.
The efficacious results from the present study of patients with lamivudine-resistant disease, a previous study on treatment-naïve patients,[12] and an in vitro study[8] suggest that LB80380 may be a good new antiviral agent in the armamentarium of the treatment of chronic hepatitis B.
In conclusion, the results of the present study suggest that LB80380 at doses of up to 240 mg is safe, well tolerated, and effective at reducing viral load in adult patients with lamivudine-resistant chronic hepatitis B for a period of 12 weeks. LB80380 showed promising results causing profound viral suppression after only 12 weeks of therapy. Further clinical investigation focusing on long-term treatment should be performed to verify the effects and safety of this compound for the treatment of chronic hepatitis B viral infection in both treatment-naïve and lamivudine-resistant patients.
Patients and Methods
Study Design and Sample Size.
This study was a phase II, open-label, multicenter, dose escalation study to evaluate the antiviral activity and safety of LB80380 during a 12-week treatment period. This study was conducted at four centers in Hong Kong and Korea. Because the efficacy assessment was descriptive, the sample size calculation was based on the accuracy of the confidence interval (CI) for the primary efficacy parameter (mean reduction in HBV DNA at 12 weeks on the log10 scale). The accuracy was measured by the width of the 95% CI. The two-sided 95% CI for the mean was estimated with the assumption that the distribution of the reduction on the log scale is symmetric and normally distributed. Assuming a potential dropout rate of at most 25% over the treatment period of 12 weeks, a minimum of twelve patients were to be enrolled into each dose group to ensure that at least nine would be evaluable.
The treatment period was divided into two parts: an initial 4-week treatment period (part 1) during which dose escalation was assessed, followed by an 8-week extension period (part 2). The treatment period was followed by a 24-week follow-up period.
During the initial 4 weeks of the treatment period (part 1), patients received LB80380 together with lamivudine 100 mg once daily. The overlapping 4-week period of LB80380 and lamivudine was designed to minimize the risk of hepatitis flares that might occur if the therapy was switched directly to LB80380 monotherapy. Five doses of LB80380 were planned: 30 mg (group 1); 60 mg (group 2); 90 mg (group 3); 150 mg (group 4), and 240 mg (group 5).
After completion of 4-week dosing at each level of LB80380 combined with lamivudine 100 mg in part 1, patients were given only LB80380 at the same dose for an additional 8 weeks in part 2, unless more than two patients in the group experienced dose-limiting toxicity (DLT) during part 1. DLT was defined as the occurrence of one of the following: grade 4 ALT (>10 x baseline) with evidence of hepatic decompensation; prothrombin time >3 seconds prolonged relative to the normal control; serum albumin level <30 g/L; total bilirubin level >5 x upper limit of normal (ULN); serum creatinine >6 x ULN or amylase value >5 x ULN; any other grade 4 clinical or laboratory toxicity considered to be reasonably or possibly related to the study drug; or any clinical and laboratory adverse events of any intensity considered to be reasonably or possibly related to study drug and necessitating permanent discontinuation of study drug.
The laboratory measures performed at every study visit for safety monitoring included complete blood count, liver and renal function (urea and creatinine), serum levels of amylase (lipase if serum amylase >1.5 x ULN), lactic acid, and creatine kinase. Estimated creatinine clearance (CrCl) according to the Cockroft-Gault formulation (based on serum creatinine level, age, body mass, and sex) were calculated in all the study visits.
After the 12-week treatment period, all patients were given adefovir dipivoxil 10 mg/day for 24 weeks (follow-up period). This treatment protocol was applied in groups 2-5. (Group 1 and one patient in group 2 were recruited under an earlier protocol in which part 2 consisted of 20 weeks instead of 8 weeks of treatment with LB80380 monotherapy. Due to a change of protocol when part 2 was in progress, these patients received 9-16 weeks of treatment with LB80380 alone instead of the 20 weeks originally planned.)
Patients visited the study sites for assessment of safety and antiviral activity at weeks 1, 2, 3, and 4 during part 1 and at weeks 8 and 12 during part 2. Thereafter, patients attended six follow-up visits at weeks 16, 20, 24, 28, 32, and 36. All available safety and tolerability data were reviewed before dose escalation. All patients within each group had to complete part 1 of the treatment period before enrollment at the next planned dose could begin. Dose escalation to the subsequent group could only be initiated if fewer than three patients experienced DLT within a given dose level during part 1 of the treatment period. Furthermore, if more than two cumulative patients within a group experienced DLT over the entire treatment period including part 1 and part 2, then further dose escalation would not be initiated.
The study was approved by the institutional review boards in all the study centers in Hong Kong and Korea. It was conducted in accordance with the study protocol and in compliance with current International Conference on Harmonisation/Good Clinical Practice guidelines, the ethical principles stated in the 1964 Declaration of Helsinki and subsequent revisions (including the 2000 Edinburgh Revision), and other applicable international and regional regulatory requirements. Informed written consent was obtained from all the patients.
Inclusion and Exclusion Criteria.
The present study recruited patients with the following criteria: age 18-65 years; presence of serum HBV surface antigen (HBsAg) for at least 6 months; and presence of hepatitis B e antigen (HBeAg) for more than 1 month with compensated liver disease. All patients had lamivudine resistance due to YMDD mutant HBV with documented detection of the HBV mutants rtM204V or rtM204I with or without the upstream mutant of rtL180M by line probe assay (INNO-LiPA HBV DR1; Innogenetics, Ghent, Belgium). All had received continuous lamivudine treatment for 6 months or more. Other inclusion criteria were: HBV DNA levels 1 x 106 copies/mL (measured by the COBAS Amplicor HBV Monitor assay; Roche Diagnostics, Branchburg, NJ; lower limit of detection 300 copies/mL); ALT value between 1.5 and 10 x ULN; and for women of child-bearing potential, negative serum pregnancy test prior to study entry, and willingness to use at least two contraception methods including a barrier method.
Patients with the following criteria were excluded: coinfection with hepatitis C, hepatitis D, and human immunodeficiency virus; pregnancy or breast-feeding; use of known nephrotoxic or hepatotoxic agents; treatment with immunomodulatory agents or corticosteroids within 6 months prior to study entry; decompensated liver disease with clinical complications of cirrhosis; prothrombin time >3 seconds prolonged relative to the normal control; serum albumin <30 g/L and bilirubin >2.5 x ULN; or other laboratory parameters, including hemoglobin <9.0 g/dL (unless due to haemoglobinopathy), absolute neutrophil count <1.5 x 109/L, platelet count <100 x 109/L, creatinine >133 mol/L, serum amylase >1.5 x ULN, lipase >1.5 x ULN, alpha-fetoprotein level >20 ng/mL, and ultrasonography performed prior to baseline with findings indicative of hepatocellular carcinoma.
Efficacy and Safety Evaluations.
The efficacy variable was serum HBV DNA levels. The primary efficacy endpoint was a reduction in log10 serum HBV DNA level from baseline at week 12. Secondary efficacy endpoints included: reduction in log10 serum HBV DNA level from baseline at week 4; proportion of patients with HBeAg seroconversion at week 12; proportion of patients with HBsAg seroconversion at week 12; and proportion of patients with ALT normalization at week 12. Evaluation of safety of the study drug was based on adverse event (AE) and serious AE data, DLT data, clinical laboratory reports, physical examinations, and vital signs. The predetermined amount of creatinine increase was set as >125% of the baseline creatinine level, and this was for easy alertness of possible abnormal data to the investigators.
HBeAg seroconversion was defined as loss of HBeAg with the development of antibody to HBeAg. Virologic rebound was defined as an increase of HBV DNA level by more than 1 log compared with the nadir in patients who achieved more than 1 log reduction of HBV DNA during the treatment period compared with baseline HBV DNA levels.
Surveillance of possible LB80380 and adefovir viral mutations were not conducted because of the limited duration of LB80380 treatment of 12 weeks and adefovir treatment for 24 weeks.
Populations for Analysis.
Evaluation of patient disposition was based on the enrolled population. The per-protocol (PP) population included all patients who were treated for at least 12 weeks with LB80380 with at least 80% compliance and had no major protocol violations. The intention-to-treat population included all patients who received at least one dose of the study medication and had serum HBV DNA values at baseline and for at least one other visit during the 12-week treatment period. Demographic and subsequent analyses were based on the PP population. Drug safety was monitored in all patients who received at least one dose of study medication.
Patient distribution and the components of the populations of analysis are illustrated in Fig. 1. One patient from group 2 was not included in the intention-to-treat population because this patient was withdrawn from the study on day 6 (due to a screening ALT >400 U/L) and consequently there were no postbaseline data for the analysis of efficacy for this patient. Four patients were excluded from the PP population due to protocol deviations. These include the group 2 patient mentioned above, one group 3 patient due to a low baseline platelet count, and one group 4 patient due to a low baseline ALT level. These three patients therefore did not complete 12 weeks of treatment as required to warrant inclusion in the PP population. The fourth patient from group 5 did not fulfill the inclusion criterion that patients had to be HBeAg-positive for more than 1 month prior to the screening visit.
Statistical Analysis.
Baseline parameters of the five groups were compared using the Kruskal-Wallis test. The decrease in log10 serum HBV DNA level from baseline to week 4 and 12 was calculated as baseline log10 serum value minus week 4 and 12 log10 serum value. A positive change from baseline corresponded to a reduction in the log10 serum HBV DNA level and was indicative of an improvement. A linear regression model was used to assess dose response in the efficacy endpoint in the PP population. The regression analysis was performed using PROC REG using log10 dose as the covariate. The regression coefficient, or dose proportionality constant, was reported together with a 95% CI. For each model, the null hypothesis of dose independence was tested against the two-sided alternative using a Wald test, and a P value generated. Because these analyses are considered exploratory, no adjustment for multiple comparisons was made. The Cochran-Armitage test for trend was used to test for trend in the proportion of patients with HBeAg seroconversion, HBsAg seroconversion, and ALT normalization at week 12 across the five dose groups (30, 60, 90, 150, or 240 mg/day). The test for trend was performed using PROC FREQ with the TREND option. The changes of the mean CrCl at baseline and week 12 were compared by way of paired t test. An exact, two-sided test was conducted at the 5% significance level.
|
|
| |
| |
|
|
|