| |
New HCV Drugs EASL 2010
|
| |
| |
ONCE-DAILY NS5A INHIBITOR (BMS-790052) PLUS PEGINTERFERON-ALPHA-2A AND RIBAVIRIN PRODUCES HIGH RATES OF EXTENDED RAPID VIROLOGIC RESPONSE IN TREATMENT-NAIVE HCV-GENOTYPE 1 SUBJECTS: PHASE 2A TRIAL
S. Pol1, G. Everson2, R. Ghalib3, V. Rustgi4, C. Martorell5, H.A. Tatum6, J. Lim7, C. Hezode8, U. Diva9, P.D. Yin9, R. Hindes9
1Hopital Cochin, Paris, France, 2University of Colorado Denver & Hospital, Denver, CO, 3The Liver Institute at Methodist Dallas Medical Center, Dallas, TX, 4Metropolitan Research, Fairfax, VA, 5The Research Institute, Springfield, MA, 6Options Health Research, Tulsa, OK, 7Yale University School of Medicine, New Haven, CT, USA, 8CHU Henri Mondor, Creteil, France, 9Bristol-Myers Squibb Company, Wallingford, CT, USA. *stanislas.pol@cch.aphp.fr
Background: BMS-790052 is a first-in-class, highly potent, once-daily HCV NS5A inhibitor. In Phase I studies in HCV-infected subjects, BMS-790052 was well-tolerated and exhibited potent antiviral activity.
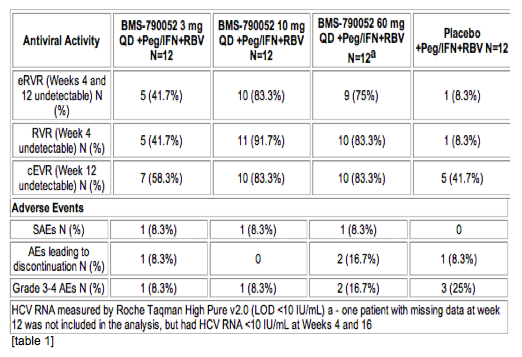
Methods: In this double-blind study, 48 subjects were randomized 1:1:1:1 to receive placebo, 3 mg, 10 mg or 60 mg of BMS-790052, once-daily in combination with peginterferon-alpha-2a and ribavirin (P/R) for 48 weeks in treatment-naive HCV genotype 1-infected subjects. The primary endpoint was the proportion of subjects with extended rapid virologic response (eRVR) defined as HCV RNA < 10 IU/mL at both Weeks 4 and 12.
Results: Subject baseline and demographic characteristics were well-balanced across treatment arms (n = 12/arm), with mean baseline HCV RNA 6.5 log10 IU/mL. The proportion of subjects achieving eRVR was 42%, 83% and 75% in the 3 mg, 10 mg and 60 mg BMS-790052 + P/R arms, respectively, compared to 8% for P/R. Safety was comparable across treatment arms (see table). Adverse events were consistent with those commonly observed with P/R. Confirmed viral breakthrough was not observed in the 10 mg and 60 mg BMS-790052 arms through Week 12.
Conclusions: BMS-790052 is a potent once-daily NS5A inhibitor that yielded higher eRVR, RVR, and cEVR rates when combined with P/R than P/R alone. The addition of BMS-790052 to P/R was well-tolerated with an AE profile comparable to P/R. These Results support further development of BMS-790052 in combination with P/R or other HCV antivirals.
Impact of Low Dose Ritonavir Boosting on the Pharmacokinetics of RG7227
(ITMN-191), a Highly Potent and Selective Inhibitor of the HCV NS3/4A Protease
Authors: J.O. Haznedar1, J. Fretland2, G. Leong2, S. Blotner3, T Hill4, P. Smith1 and J.Q. Tran1
1Roche, Clinical Pharmacology, South San Francisco, CA, USA;
2Roche Palo Alto LLC, Department of Drug Metabolism and Pharmacokinetics, Palo Alto, CA, USA;
3Hoffmann-La Roche Inc., Pharma Development Innovation Biomathematics, Nutley, NJ, USA;
4Hoffmann-La Roche Inc., Clinical Research & Exploratory Development Operations, Nutley, NJ, USA
Background: Low dose ritonavir (RTV), a potent CYP3A inhibitor, is being used to "boost" the pharmacokinetics (PK) of HIV protease inhibitors (PIs) which are CYP3A substrates, enabling regimen simplification while improving treatment response. RG7227, an HCV NS3/4A PI, is a CYP3A substrate and RTV has a potential to enhance its PK. This Phase 1 study investigated the effects of low dose RTV on the single-dose PK of RG7227.
Material & Methods: A single-center, open-label Phase 1 study in 14 healthy males and females between 18 and 45 years of age. Subjects received RTV 100 mg twice-daily (BID) for 10 days. Single oral doses of RG7227 100 mg were administered on 3 separate occasions: alone (2 days before ritonavir dosing), with the first dose of RTV, and on the last day of RTV dosing. Serial PK blood samples for the measurement of RG7227 were collected at pre-dose and up to 48 hours post-dose.
Results: Twelve subjects completed the study. Single doses of RTV 100 mg increased RG7227 AUC0-inf, Cmax and C12h by 5.69-fold (90% CI: 4.06, 7.96), 3.14-fold (90% CI: 1.87, 5.25) and 50.6-fold (90% CI: 25.6, 100), respectively. Multiple doses of RTV 100 mg BID for 10 days had similar effects on RG7227 AUC0-inf (5.46-fold; 90% CI: 4.03, 7.40) and Cmax (3.19-fold; 90% CI: 2.09, 4.85). The multiple-dose effect of ritonavir on RG7227 C12h was less pronounced than the single dose ritonavir effect (26.9-fold; 90% CI: 17.7, 41.0). This is likely due to the mixed inhibition and induction effects of ritonavir on CYP enzymes. No new safety findings were observed.
Conclusion: Low dose RTV significantly enhances RG7227 Cmin, which appears to be the key parameter driving efficacy and prevention of resistance. Less of an effect was observed on AUC and Cmax, which are parameters often attributed to safety. RG7227 dose and dosing frequency may be reduced when co-administered with RTV, to achieve a high Cmin while providing similar or lower AUC and Cmax compared to an unboosted regimen. A Phase 1b study of RTV-boosted RG7227 QD and BID regimens in combination with SOC in HCV-infected patients is being conducted.
Ritonavir Boosting of Low Dose RG7227/ITMN-191, HCV NS3/4A Protease Inhibitor, Results in Robust Reduction in HCV RNA at Lower Exposures Than Provided by Unboosted Regimens
Authors: E. Gane1, R. Rouzier2, C. Stedman3, A. Wiercinska-Drapalo4, A. Horban5, L. Chang6, Y. Zhang6, P. Sampeur6, I. Najera6, N. Shulman6, P. Smith6 and J. Tran6
1. Auckland Clinical Studies, New Zealand
2. CENTRE CAP, France
3. Christchurch Clinical Studies Trust, New Zealand
4. Department of Hepatology and Immunodeficiencies, Warsaw Medical University, Poland
5. Warsaw Medical University &Hospital of Infectious Diseases. Warsaw, Poland
6. Roche, USA
Background: Co-administration of low dose ritonavir resulted in larger increases in RG7227 trough concentration than area under the curve (AUC) and marginal increase in maximum concentration (Cmax), offering the potential to reduce the dose and/or dosing frequency of RG7227 in future Phase 2 studies. The present study evaluated the safety, tolerability, antiviral activity and pharmacokinetics (PK) of once-daily (QD) and twice-daily (BID) ritonavir boosted RG7227 (RG7227/r) in combination with standard of care (SOC) in treatment-naïve HCV genotype 1 patients.
Material & Methods: In a double-blind, placebo-controlled, Phase 1b multiple-ascending dose (MAD) study, treatment-naïve HCV genotype 1 patients were randomized to receive RG7227/r or placebo/r plus SOC for 15 days. RG7227/r regimens were 100/100mg BID (Cohort 1), 200/100mg QD (Cohort 2), and 200/100mg BID (Cohort 3). Blood samples for HCV RNA, PK, and resistance monitoring were collected at baseline and throughout the study.
Results: Cohort 1 and 2 provided blinded data for this abstract. The following table summarizes virologic response of RG7227/r compared to un-boosted 900 mg BID, the highest dose evaluated in a previous MAD study (placebo/r data are not shown due to small N of 2):
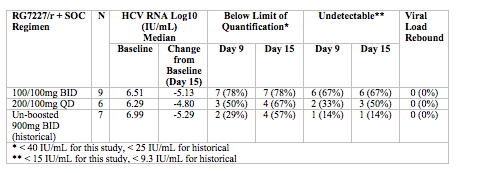
RG7227/r AUC and Cmax were approximately 6- to 9-fold and 4- to 10-fold lower, respectively, than previously observed for 900 mg BID un-boosted. Inter-patient PK variability for RG7227/r was also lower than historical un-boosted RG7227. RG7227/r and placebo/r plus SOC were generally well tolerated. Complete data for all 3 cohorts will be presented.
Conclusion: Relative to un-boosted 900 mg BID, ritonavir boosting of low dose RG7227 provides robust and sustained virologic response at significantly lower AUC and Cmax, parameters which are often correlated with safety. These Results indicate that further exploration of ritonavir boosting on the safety and efficacy of RG7227 is warranted.
SVR WITH TELAPREVIR, PEGINTERFERON ALFA-2A AND RIBAVIRIN IN HCV PATIENTS WITH WELL-CHARACTERIZED PRIOR NULL RESPONSE, PARTIAL RESPONSE, VIRAL BREAKTHROUGH OR RELAPSE AFTER PR
T. Berg1, J.G. McHutchison2, N. Adda3, F. Poordad4, M.L. Shiffman5, P. Ferenci6, J. Heathcote7, J.-M. Pawlotsky8, S. Zeuzem9, H. Reesink10, G. Dusheiko11, E. Martin3, D. Alexanderian3, S. George3, A.J. Muir2
1University Clinic, Leipzig, Leipzig, Germany, 2Duke University Medical Center, Durham, NC, 3Vertex Pharmaceuticals Incorporated, Cambridge, MA, 4Cedars-Sinai Medical Center, Los Angeles, CA, 5Bon Secours Health System, Liver Institute of Virginia, Newport News, VA, USA, 6University of Vienna, Vienna, Austria, 7University of Toronto, Toronto, ON, Canada, 8Hopital Henri Mondor, Creteil, France, 9Johann Wolfgang Goethe University Medical Center, Frankfurt/Main, Germany, 10Academisch Medical Center, University of Amsterdam, Amsterdam, The Netherlands, 11Royal Free and University College School of Medicine, London, UK. *thomas.berg@charite.de
Background and aims: Study107 is an open-label rollover study of telaprevir(T) with peginterferon+ribavirin(PR) in genotype-1 HCV patients who did not achieve SVR following PR treatment in telaprevir Phase 2 studies.
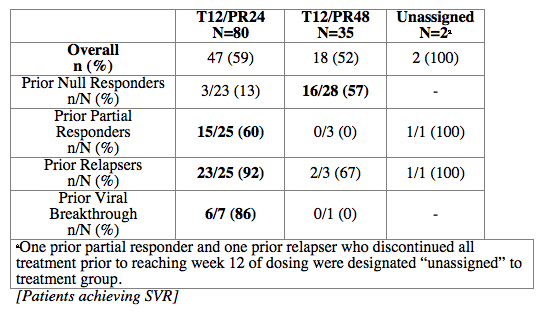
Methods: Null responders (< 1-log10 HCV RNA decrease at week-4 or < 2-log10 at week-12), partial responders (≥2-log10 decrease at week-12, detectable at week-24), patients with viral breakthrough and relapsers from PROVE1/2/3 PR arms were eligible for treatment. Initially, all patients received T 750mg q8h plus PR at standard doses for 12 weeks, followed by 12 weeks of PR(T12/PR24). Protocol was amended to allow partial responders, viral breakthroughs and relapsers with undetectable HCV RNA at weeks 4 and 12 (eRVR) to receive T12/PR24. Partial responders, viral breakthroughs and relapsers with detectable HCV RNA at week-4 and/or week-12 and null responders received an additional 24 weeks of PR(T12/PR48).
Results: Of 117 patients included in an ITT analysis, 97(83%) had baseline HCV RNA ≥800,000IU/mL, 69(59%) had genotype subtype 1a, 44(38%) had cirrhosis or bridging fibrosis, and 9(8%) were black. Viral breakthrough and relapse rates occured in 25%, 23% of prior null responders; 10%, 22% of prior partial responders; 13%, 0% of prior viral breakthroughs; and 0%, 4% of prior relapsers.
The most frequent AEs (≥20%) were fatigue, flu-like-syndrome, nausea, diarrhea, pruritus, rash, headache, insomnia and anemia. Grade 3 rash and Grade 3 anemia were observed in 6(5%) and 6(5%) patients, respectively. Ten(9%) patients discontinued due to AEs, 5(4%) due to rash and 2(2%) to anemia.
Conclusions: Patients with prior relapse, breakthrough and partial response exhibited high SVR rates after 24 weeks of telaprevir-based regimen. High SVR rates were also observed in patients with prior null response after 48 weeks of therapy.
MK-5172: A NOVEL HCV NS3/4A PROTEASE INHIBITOR WITH POTENT ACTIVITY AGAINST KNOWN RESISTANCE MUTANTS
S. Carroll1, J. McCauley1, S. Ludmerer1, S. Harper2, V. Summa2, M. Rowley2, M. Rudd1, P. Coleman1, N. Liverton1, J. Butcher1, C. Mcintyre1, J. Romano1, K. Bush1, M. Ferrara2, B. Crescenzi2, A. Petrocchi2, M. Difilippo2, C. Burlein1, J. Dimuzio1, D. Graham1, C. Mchale1, M. Stahlhut1, A. Gates1, C. Fandozzi1, N. Trainor1, D. Hazuda1, J. Vacca1, D. Olsen1
1Antiviral Research, 2Medicinal Chemistry, Merck Research Laboratories, West Point, PA, USA, 3Department of Antiviral Research, Instituto di Ricerche di Biologia Molecolare, Pomezia, Italy, 4Drug Metabolism, Merck Research Laboratories, West Point, PA, USA. *nigel_liverton@merck.com
Background and aims: Efforts toward novel HCV treatment options include the development of direct antiviral agents which inhibit key steps in viral replication, such as HCV NS3/4a protease. Several inhibitors of HCV NS3/4A protease activity have demonstrated antiviral efficacy in clinical studies. However viral variants resistant to inhibition by these compounds have been detected in patients. Our interest is in identifying HCV NS3/4a protease inhibitors displaying good activity against known resistant viral variants that will result in low rates of treatment failure due to viral resistance.
Methods: Medicinal chemistry efforts focused on a novel macrocyclic scaffold and were guided by biochemical and cell-based replicon assays that utilized forms of HCV NS3/4a protease that included known resistance mutations R155K, D168V, and A156T. In vivo efficacy was evaluated in chimpanzees harboring chronic HCV infections.
Results: MK-5172 was identified as a potent macrocyclic inhibitor of NS3/4a (genotype 1b enzyme Ki ∼ 0.02 nM, genotype 1b replicon IC50 = 3.5 nM). In both biochemical and cell-based replicon assays, MK-5172 retained significant inhibitory potency against NS3/4A variants R155K (Ki = 0.07 nM), D168V (Ki 0.3 nM), and A156T (Ki = 5 nM). MK-5172 exhibits significant liver exposure in rat, following a 5 mg/kg oral dose ([rat liver]4h = 27 µM) thus demonstrating effective distribution of compound into liver, the site of HCV replication. Three HCV-infected chimpanzees infected with either wild-type or R155K virus were dosed with MK-5172 orally for seven days at 1 mg/kg b.i.d. All demonstrated high concentrations of MK-5172 in the liver; wild-type virus experienced rapid suppression of greater than three logs, while R155K virus experienced an immediate 1-2 log reduction.
Conclusions: MK-5172 is a novel NS3/4a protease inhibitor with an attractive preclinical profile including high in vitro potency, significant liver exposure upon oral dosing, and potent inhibitory activity against known viral variants that are resistant to other protease inhibitors in development. MK-5172 has also demonstrated antiviral efficacy in a chimpanzee model of chronic HCV infection. Clinical studies with MK-5172 are currently in progress.
SAFETY AND ANTIVIRAL ACTIVITY OF THE HCV NON-NUCLEOSIDE POLYMERASE INHIBITOR VX-222 IN TREATMENT-NAïVE GENOTYPE 1 HCV-INFECTED PATIENTS
M. Rodriguez-Torres1, E. Lawitz2, B. Conway3, K. Kaita4, A.M. Sheikh5, R. Ghalib6, R. Adrover7, C. Cooper8, M. Silva7, M. Rosario9, B. Bourgault10, L. Proulx10, J.G. McHutchison11
1Fundacion de Investigacion de Diego, Santurce, PR, 2Alamo Medical Research, San Antonio, TX, USA, 3University of British Columbia, Vancouver, BC, 4University of Manitoba, Winnipeg, MB, Canada, 5GI Specialists of Georgia, Marietta, GA, 6The Liver Institute at Methodist Dallas, Dallas, TX, USA, 7ACLIRES, Buenos Aires, Argentina, 8University of Ottawa, Ottawa, ON, Canada, 9Hospital Universitario Austral, Buenos Aires, Argentina, 10Vertex Pharmaceuticals Incorporated, Cambridge, MA, USA, 11ViroChem Pharma Inc., Laval, QC, Canada, 12Duke University Medical Center, Durham, NC, USA. *rodztorres@coqui.net
Background and aims: VX-222 is a novel non-nucleoside hepatitis C virus (HCV) polymerase inhibitor with potent in vitro activity. Safety, tolerability, pharmacokinetics and antiviral activity of VX-222 were assessed in a phase Ib/IIa multicenter, randomized, double-blinded, placebo-controlled, dose-ascending study in HCV-infected patients.
Methods: Treatment-naïve HCV genotype 1 patients were randomized to receive VX-222 at doses of 250 mg BID, 500 mg BID, 750 mg BID, 1500 mg QD or placebo for 3 days in a treatment:placebo ratio of 6:2 (8 patients/cohort).
Peginterferon-alfa-2a (P) plus ribavirin (R) was offered to patients at end of study (day 4) for up to 48 weeks, as judged appropriate by the investigator. PR treatment was discontinued in patients who had not experienced a ≥2 log10 HCV RNA level decline at week 12. VX-222 plasma levels were assessed at multiple time points over 12 hours, on days 1 and 3.
Results: Twenty-four patients were enrolled in the first three cohorts. VX-222 exposure increased in a dose-related manner. On day 3, the mean AUC0-12h and Cmax values were 19,490 ng*h/mL (CV 41%) and 2,959 ng/mL (CV 29%), 29,848 ng*h/mL (CV 54%) and 5,044 ng/mL (CV 36%), and 62,952 ng*h/mL (CV 112 %) and 10,288 ng/mL (CV 112%) for the 250 mg, 500 mg, and 750 mg BID groups, respectively. The mean HCV RNA decline achieved on day 4 with placebo, 250 mg, 500 mg, and 750 mg VX-222 BID was 0.1 log10 (range: 0.3 increase to 0.5 decline), 3.1 log10 (range: 2.0 to 4.2), 3.4 log10 (range: 3.2 to 3.6), and 3.2 log10 (range: 2.3 to 3.8), respectively. All AEs reported were mild to moderate, and the most frequently reported AEs by patients that received either active drug or placebo were diarrhea (25%), headache (20%) and nausea (12%). No clinically relevant laboratory abnormalities were reported.
Conclusions: VX-222 was well tolerated and a mean HCV RNA decline of >3 log10 at day 4 was observed in each cohort, Results of the fourth cohort evaluating QD dosing will also be presented. These Results support further evaluation of VX-222 in combination with peginterferon and ribavirin in the treatment of HCV.
IDENTIFICATION AND CHARACTERIZATION OF PPI-461, A POTENT AND SELECTIVE HCV NS5A INHIBITOR WITH ACTIVITY AGAINST ALL HCV GENOTYPES
R. Colonno, E. Peng, M. Bencsik, N. Huang, M. Zhong, A. Huq, Q. Huang, J. Williams, L. Li
Presidio Pharmaceuticals, San Francisco, CA, USA. *rich@presidiopharma.com
Background: Several mechanistic classes of oral HCV inhibitors are currently undergoing clinical development. Inhibitors of NS5A have distinguished themselves by their picomolar potency and broad genotype spectrum in HCV replicon assays. Here we report the discovery and preclinical profile of PPI-461, a newly discovered NS5A inhibitor being advanced toward clinical studies.
Methods: Antiviral potency, combination and resistance studies utilized standard HCV replicon systems (genotype 1a and 1b). HCV spectrum studies employed stable or transient replicon assays with insertion of NS5A gene segments from other HCV genotypes (2a, 3a, 4a, 5a, 6a and 7a) into an HCV 1b backbone. Pharmacokinetic studies utilized IV and PO administration of PPI-461 in rats, monkeys and dogs. Extensive ADME and toxicology profiling has been performed in vitro and in rats and monkeys.
Results: PPI-461 was identified through an extensive medicinal chemistry effort and exhibits EC50s of 0.2 and 0.01 nM in HCV 1a and 1b replicon assays, respectively. Antiviral activity (EC50s of 0.1-19 nM) is maintained against all other HCV genotypes in transient/stable replicon assays. Cellular cytotoxicity levels (CC50) are >10 uM in several cell lines and no activity is observed against several other viruses (including BVDV) at 10 uM. PPI-461 is additive to synergistic (CIs 0.29-0.89) when combined with IFN, NS3 protease, NS5B nucleoside and non-nucleoside inhibitors. Resistance studies showed that variants with high levels of resistance could be generated, especially with genotype 1a, but required multiple amino acid substitutions within the NS5A gene. PPI-461 shows excellent stability in liver microsomes and no significant inhibition against P450 isozymes at 10 uM. PPI-461 exhibits good oral bioavailability in rats, monkeys and dogs, with enhanced liver concentrations and plasma half-lives suggestive of once daily dosing in humans. Toxicology studies in rats and monkeys showed that PPI-461 is well tolerated at plasma exposure levels >100,000-fold HCV 1a EC50 levels.
Conclusion: HCV NS5A protein plays a critical role in the HCV viral replicative cycle and is an attractive target for antiviral intervention. PPI-461 is a newly discovered selective and broadly potent inhibitor of HCV NS5A protein with a favorable preclinical profile supportive of advancement into clinical trial
ILIBININ AS A RESCUE TREATMENT FOR HCV-INFECTED PATIENTS SHOWING SUBOPTIMAL VIROLOGIC RESPONSE TO STANDARD COMBINATION THERAPY
M. Biermer, L. Stoehr, B. Schlosser, B. Fulop, F. van Bommel, T. Berg
Medizinische Klinik mit Schwerpunkt Gastroenterologie und Hepatologie, Charite Campus Virchow, Berlin, Germany. *michael.biermer@charite.de
During standard antiviral treatment a significant proportion of hepatitis C patients show no complete virologic response despite a marked reduction of viral load in early stages of treatment (partial response). This minimal viremia may persist indicating the treatment termination as a consequence of viral nonresponse.
Recently, high-dose intravenous silibinin has demonstrated significant antiviral activity. We were therefore interested whether patients with partial virologic response can be rescued by the on-treatment addition of high-dose intravenous silibinin infusions.
Eleven patients who received different interferon-based antiviral regimens qualified for the rescue strategy and received silibinin infusions between December 2008 and September 2009. All patients had received peginterferon alfa 2a / ribavirin and four patients additionally received an HCV specific protease inhibitor for the first four weeks of treatment. 9 out of 11 patients showed a plateau of HCV RNA levels ranging from detectable but not quantifiable (< 15 IU/mL) to 2015 IU/mL for at least eight weeks on continuous treatment (total treatment duration 16-30 weeks). The remaining two patients suffered a virologic rebound after cessation of the protease inhibitor (HCV RNA 484 IU/ml at week 6 (patient 1) and 6890 IU/ml at week 5 (patient 2). Every patient received silibinin infusions (1400 mg/day - in 500 ml NaCl) on two consecutive days.
Eight out of eleven patients achieved a complete suppression of viral replication below the limit of detection within the first week after silibinin. Three patients did initially not respond to silibinin, two of the responders showed a viral breakthrough (8 and 14 weeks after silibinin administration during continued SOC treatment) and up to now six patients have undetectable HCV-RNA levels eight to 40 weeks after silibinin administration with continued SOC. Silibinin infusions were generally well tolerated. All patients reported a transient sensation of heat, three patients reported painful bowel-movements, two with a single episode of vomiting.
A two days administration of high-dose intravenous silibinin might be an interesting approach to rescue patients with ongoing minimal residual viremia while on interferon-based therapy. Further follow-up is needed to confirm whether the response induced is long lasting and leads to SVR.
RITONAVIR BOOSTING OF LOW DOSE RG7227/ITMN-191, HCV NS3/4A PROTEASE INHIBITOR, Results IN ROBUST REDUCTION IN HCV RNA AT LOWER EXPOSURES THAN PROVIDED BY UNBOOSTED REGIMENS
E. Gane1, R. Rouzier2, C. Stedman3, A. Wiercinska-Drapalo4, A. Horban4, L. Chang5, Y. Zhang5, P. Sampeur6, I. Najera7, N. Shulman7, P. Smith5, J. Tran5
1Auckland Clinical Studies, Auckland, New Zealand, 2Centre CAP, Montpellier, France, 3Christchurch Clinical Studies Trust, Christchurch, New Zealand, 4Department of Hepatology and Immunodeficiencies, Warsaw Medical University, 5Warsaw Medical University & Hospital of Infectious Diseases, Warsaw, Poland, 6Roche, South San Francisco, CA, 7Roche, Nutley, NJ, 8Roche, Palo Alto, CA, USA. *edgane@adhb.govt.nz
Background: Co-administration of low dose ritonavir resulted in larger increases in RG7227 trough concentration than area under the curve (AUC) and marginal increase in maximum concentration (Cmax), offering the opportunity to reduce the dose and/or dosing frequency of RG7227 in future Phase 2 studies. The present study evaluated the safety, tolerability, antiviral activity and pharmacokinetics (PK) of once-daily (QD) and twice-daily (BID) ritonavir boosted RG7227 (RG7227/r) in combination with standard of care (SOC) in treatment-naïve HCV genotype 1 patients.
Material & Methods: In a double-blind, placebo-controlled, Phase 1b multiple-ascending dose (MAD) study, treatment-naïve HCV genotype 1 patients were randomized to receive RG7227/r or placebo/r plus SOC for 15 days. RG7227/r regimens were 100/100mg BID (Cohort 1), 200/100mg QD (Cohort 2), and 200/100mg BID (Cohort 3). Blood samples for HCV RNA, PK, and resistance monitoring were collected at baseline and throughout the study.
Results: Cohort 1 and 2 provided blinded data for this abstract. The following table summarizes virologic response of RG7227/r compared to un-boosted 900 mg BID, the highest dose evaluated in a previous MAD study (placebo/r data are not shown due to small N of 2):
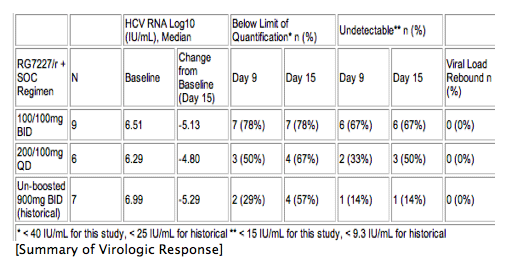
RG7227/r AUC and Cmax were approximately 6- to 9-fold and 4- to 10-fold lower, respectively, than previously observed for 900 mg BID un-boosted. Inter-patient PK variability for RG7227/r was also lower than historical un-boosted RG7227. RG7227/r and placebo/r plus SOC were generally well tolerated. Complete data for all 3 cohorts will be presented.
Conclusion: Relative to un-boosted 900 mg BID, ritonavir boosting of low dose RG7227 provides robust and sustained virologic response at significantly lower AUC and Cmax, parameters which are often correlated with safety. These Results indicate that further exploration of ritonavir boosting on the safety and efficacy of RG7227 is warranted.
VITAMIN D SUPPLEMENT IMPROVE SVR IN CHRONIC HEPATITIS C (GENOTYPE 1) NAïVE PATIENTS TREATED WITH PEG INTERFERON AND RIBAVIRIN
S. Abu Mouch1,2, Z. Fireman1,2, J. Jarchovsky1,2, N. Assy2,3
1Hepatology Unite, 2Internal Medicine B, Hillel Yaffe Medical Center, Hadera, 3Faculty of Medicine, Technion, Haifa, 4Gastroenterology, Hillel Yaffe Medical Center, Hadera, 5Liver Unit, Ziv Medical Center, Safed, Israel. *saif@hy.health.gov.il
Background: The combination therapy of Peg/RBV is considered the standard of care for chronic hepatitis C (HCV). A sustained viral response (SVR) is obtained in 40-50% of naïve HCV patients with genotype 1. Vitamin D is a potent immuno modulator whose impact on SVR of Peg/RBV based treatment of chronic HCV is unknown.
Aim: To assess whether the supplement of vitamin D to the conventional Peg/RBV therapy could improve SVR.
Methods: Fifty-eight patients with chronic HCV infection were randomized into two groups (intent-to-treat population): 27 (treatment group, age 47±11 yrs, body mass index [BMI] 27±4, 50% male) received pegylated-interferon-alpha2b (1.5 µg/kg once weekly) plus ribavirin (1000-1200 mg/daily) together with vitamin D3 (1000-4000 IU/daily, serum level >32 ng/ml), and 31 (controls, age 49±7 years, BMI 24±3, 60% male) received the same therapy without vitamin D. HCV RNA was assessed by RT-PCR (sensitivity, 50 IU/mL). Undetectable HCV RNA at week 12 and at week 24 post treatment (considered as complete EVR and SVR respectively).
Results: Demographics, disease characteristics, ethnicity, baseline biochemical parameters and adherence to treatment were similar in both groups. The treatment group had a higher mean BMI (27±4 vs 24±3; P< 0.01), viral load (68% vs 58%, P< 0.01), and fibrosis (Metavir scores >F2: 55% vs 18%, P< 0.001) than the controls. All but one treated patient (96%) and 48% (15/31) controls were HCV-RNA negative at week 12 (P< 0.0001). At week 24 post treatment (SVR): 86% (13/15) of treated patients and 41% (5/12) of controls were HCVRNA negative (P< 0.001). AEs were mostly mild and typical of Peg/RBV. There were no SAE.
Conclusions: Supplement of Vitamin D to conventional Peg/RBV therapy for naïve, genotype 1 patients with chronic infection significantly improve SVR.
SILEN-C2: EARLY ANTIVIRAL ACTIVITY AND SAFETY OF BI 201335 COMBINED WITH PEGINTERFERON ALFA-2A AND RIBAVIRIN (PEGIFN/RBV) IN CHRONIC HCV GENOTYPE-1 PATIENTS WITH NON-RESPONSE TO PEGIFN/RBV
M. Sulkowski1, M. Bourliere2, J.-P. Bronowicki3, A. Streinu-Cercel4, L. Preotescu4, T. Asselah5, J.-M. Pawlotsky6, S. Shafran7, S. Pol8, F.A. Caruntu4, S. Mauss9, D. Larrey10, C. Hafner11, Y. Datsenko11, J. Stern12, R. Kubiak11, G. Steinmann11
1Department of Viral Hepatitis, Johns Hopkins University, Baltimore, MD, USA, 2Hopital Saint Joseph, Marseille, 3Hopital de Brabois, Vandoeuvre Cedex, France, 4'Prof. Dr. Matei Bals' Institute of Infectious Diseases 1, Bucharest, Romania, 5Hopital Beaujon, Clichy Cedex, 6Hopital Henri Mondor, Creteil, France, 7University of Alberta, Edmonton, AB, Canada, 8Hopital Cochin, Paris, France, 9Center for HIV and Hepatogastroenterology, Dusseldorf, Germany, 10Hopital Saint-Eloi, Montpellier Cedex, France, 11Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach/ Ri, Germany, 12Boehringer Ingelheim Pharmaceuticals, Ridgefield, CT, USA. *msulkowski@jhmi.edu
Background and aims: BI 201335 is a potent HCV NS3/4A protease inhibitor being studied in phase IIb trials of chronic HCV genotype-1 (GT1) infection.
Methods: In a double-blind, randomized, parallel group design, HCV GT1 patients with confirmed non-response to at least 12 wks of PegIFN/RBV treatment were randomized 1:2:1 to (1) 240 mg BI 201335 once daily (QD), (2) 240 mg BI 201335 QD after a 3 day lead-in phase (LI) of PegIFN/RBV, and (3) 240 mg BI 201335 twice daily (BID) after a 3 day LI. Relapsers and patients with liver cirrhosis were excluded. In each group, treatment is for 24 wks with a Background of PegIFN (180 µg/wk) and RBV (1000/1200mg/d). Pre-specified Interim analysis after 12 weeks of therapy is reported. Viral rebound is defined as an increase in plasma HCV RNA ≥ 1 log10 on-treatment from nadir or confirmed increase ≥ 100 IU/ml if previously undetectable.
Results: 288 patients were treated (mean age 49 +/- 9 years; mean BMI 26.4 +/- 4.4 kg/m2; mean log10 HCV RNA at baseline 6.6 IU/mL). BI 201335 with PegIFN/RBV was overall well tolerated and demonstrated potent antiviral activity in all dose groups (Table). Mean ALT improved in all groups. 8% of patients prematurely discontinued treatment due to adverse events (AE). Most frequent AE were gastrointestinal disorders, mostly mild jaundice resulting from isolated unconjugated hyperbilirubinemia, (9.2%, 14.1% and 34.3% in groups 1, 2 and 3) and mostly mild to moderate rash or photosensitivity reactions (severe rash in 1.3, 0.7 and 5.7% in groups 1, 2 and 3).
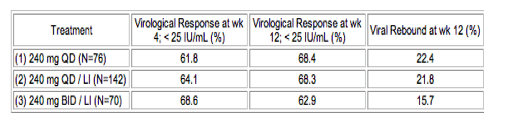
Conclusions: SILEN-C2 confirmed robust antiviral activity with overall good tolerability and safety of BI 201335 especially if given 240mg once daily in combination with PegIFN/RBV in patients with chronic HCV GT-1 infection who did not respond to a previous course of PegIFN/RBV therapy.
|
|
| |
| |
|
|
|