| |
Telaprevir for Previously Treated Chronic HCV Infection - NEJM
|
| |
| |
from Jules: next week at the liver meeting EASL in Vienna new & better data on telaprevir in null responders will be presented.
NEJM April 7 2010
John G. McHutchison, M.D., Michael P. Manns, M.D., Andrew J. Muir, M.D., Norah A. Terrault, M.D., Ira M. Jacobson, M.D., Nezam H. Afdhal, M.D., E. Jenny Heathcote, M.D., Stefan Zeuzem, M.D., Hendrik W. Reesink, M.D., Jyotsna Garg, M.S., Mohammad Bsharat, Ph.D., Shelley George, M.D., Robert S. Kauffman, M.D., Ph.D., Nathalie Adda, M.D., Adrian M. Di Bisceglie M.D., for the PROVE3 Study Team
ABSTRACT
Background - Patients with genotype 1 hepatitis C virus (HCV) who do not have a sustained response to therapy with peginterferon alfa and ribavirin have a low likelihood of success with retreatment.
Methods - We randomly assigned patients with HCV genotype 1 who had not had a sustained virologic response after peginterferon alfaĐribavirin therapy to one of four treatment groups: 115 patients to the T12PR24 group, receiving telaprevir (1125-mg loading dose, then 750 mg every 8 hours) for 12 weeks and peginterferon alfa-2a (180 µg per week) and ribavirin (1000 or 1200 mg per day, according to body weight) for 24 weeks; 113 patients to the T24PR48 group, receiving telaprevir for 24 weeks and peginterferon alfa-2a and ribavirin for 48 weeks (at the same doses as in the T12PR24 group); 111 patients to the T24P24 group, receiving telaprevir and peginterferon alfa-2a for 24 weeks (at the same doses as in the T12PR24 group); and 114 patients to the PR48 (or control) group, receiving peginterferon alfa-2a and ribavirin for 48 weeks (at the same doses as in the T12PR24 group). The primary end point was sustained virologic response (undetectable HCV RNA levels 24 weeks after the last dose of study drugs).
Results - The rates of sustained virologic response in the three telaprevir groups - 51% in the T12PR24 group, 53% in the T24PR48 group, and 24% in the T24P24 group - were significantly higher than the rate in the control group (14%; P<0.001, P<0.001, and P=0.02, respectively). Response rates were higher among patients who had previously had relapses than among nonresponders. One of the most common adverse events in the telaprevir groups was rash (overall, occurring in 51% of patients, with severe rash in 5%). Discontinuation of study drugs because of adverse events was more frequent in the telaprevir groups than in the control group (15% vs. 4%).
Conclusions - In HCV-infected patients in whom initial peginterferon alfa and ribavirin treatment failed, retreatment with telaprevir in combination with peginterferon alfa-2a and ribavirin was more effective than retreatment with peginterferon alfa-2a and ribavirin alone.
Worldwide, an estimated 170 million people have chronic hepatitis C virus (HCV) infection, and in many of these people, cirrhosis and complications of end-stage liver disease will develop.1,2 Chronic HCV infection is the leading cause of liver disease and is the leading indication for liver transplantation in the United States and Europe.3 Currently available treatment for HCV infection necessitates peginterferon alfa injections combined with oral ribavirin for 24 or 48 weeks (depending on the HCV genotype).4 Overall, in 40 to 50% of patients infected with HCV genotype 1 who have not previously been treated, the currently available treatment results in short- and longer-term benefits associated with sustained virologic response.5,6,7,8,9,10,11 For patients in whom a sustained virologic response is not achieved, retreatment options are limited to reexposure to the same medications, with potential modification of the dose or duration of the regimen. These retreatment strategies are associated with clinically significant morbidity and generally have a very limited chance of resulting in a successful outcome.11,12 Therefore, the development of effective regimens to retreat patients with chronic HCV infection who did not have a sustained response to previous therapy is an urgent priority.
Telaprevir, an orally bioavailable inhibitor of the nonstructural 3/4A (NS3/4A) HCV protease,13 is one of several investigational agents that specifically targets HCV with the goal of improving the chance of a sustained virologic response. In two recent phase 2, multicenter studies of telaprevir in combination with peginterferon alfa-2a and ribavirin in patients infected with HCV genotype 1 who had not previously been treated, the addition of telaprevir for 12 weeks, as part of a combination regimen with a total duration of 24 weeks in most patients, significantly increased the rates of sustained virologic response, albeit with more patients in the telaprevir group than in the control group discontinuing therapy because of adverse events.14,15 In the current study, we aimed to assess the safety and efficacy of telaprevir-based therapy in patients with HCV infection who had not had a sustained response to an initial full course of treatment with peginterferon alfa and ribavirin.
Methods
Study Conduct
An independent data and safety monitoring committee evaluated the rates of viral breakthrough and the safety and tolerability of the study drugs on the basis of adverse events and laboratory values during the first 24 weeks of treatment. Three prespecified, unblinded, interim reviews were conducted by the committee: one when 25% of patients had completed 4 weeks of treatment, one when all patients had completed 12 weeks of treatment, and one when 50% of patients had completed 24 weeks of treatment. After each review, the independent data and safety monitoring committee provided a summary report, without revealing the unblinded data, which was distributed to the clinical sites. The data management and interim analyses were performed by the Duke Clinical Research Institute. A single statistical interim analysis, conducted by a statistical group that was independent of the sponsor, was conducted after the last patient completed the week 36 visit. The analysis did not involve analysis of the primary end point; therefore, no adjustment to the two-sided, 5% significance level was needed.
The study sponsor, Vertex Pharmaceuticals, and the academic principal investigators were jointly responsible for study design, protocol development, study coordination, and drafting and review of previous drafts of the manuscript. A study publication committee, consisting of a majority of academic authors, agreed on a plan to submit the manuscript for publication. All authors had access to the data, vouch for the accuracy and completeness of the data and analyses, and contributed to the writing of the manuscript.
Study Patients
Patients were enrolled at 53 international sites (41 in the United States [including 1 in Puerto Rico], 6 in Canada, 3 in the Netherlands, and 3 in Germany). Eligible patients were 18 to 70 years of age, had chronic infection with HCV genotype 1, and had previously been treated for HCV infection with peginterferon alfa and ribavirin but did not have a sustained virologic response.
Patients' previous response to at least 12 weeks of therapy with peginterferon alfa and ribavirin was determined from medical records and records of previous HCV RNA levels and was defined as a nonresponse (i.e., undetectable HCV RNA levels never achieved during or at the end of the treatment period), relapse (i.e., undetectable HCV RNA levels achieved during treatment for at least 42 weeks but detectable HCV RNA levels observed during the follow-up period and lack of achievement of a sustained virologic response), or breakthrough (i.e., undetectable HCV RNA levels during the treatment period but detectable levels before the end of the treatment period).
Eligible patients were also seronegative for hepatitis B surface antigen and antibodies against human immunodeficiency virus types 1 and 2 and had an absolute neutrophil count of 1500 or more per cubic milliliter, a platelet count of 100,000 or more per cubic milliliter, and normal bilirubin values. Patients with decompensated liver disease, hepatocellular carcinoma, or other clinically significant liver disease were excluded. A liver biopsy had to have been performed within 3 years before the study. The protocol was approved by the institutional review boards for all participating countries and study centers, and the study was performed according to Good Clinical Practice Guidelines. All patients provided written informed consent before participating in the study.
Study Design
This trial was a randomized, stratified, partially placebo-controlled, partially double-blind, phase 2 study. Patients received one of four possible courses of treatment.
The T12PR24 group received telaprevir (VX-950, Vertex Pharmaceuticals) plus peginterferon alfa-2a (Pegasys, Roche) and ribavirin (Copegus, Roche) for 12 weeks, followed by placebo and peginterferon alfa-2a and ribavirin for the next 12 weeks. The T24PR48 group received telaprevir plus peginterferon alfa-2a and ribavirin for 24 weeks, followed by peginterferon alfa-2a and ribavirin for the next 24 weeks. The T24P24 group received telaprevir and peginterferon alfa-2a for 24 weeks. The PR48 (control) group received placebo plus peginterferon alfa-2a plus ribavirin for 24 weeks, followed by peginterferon alfa-2a and ribavirin for the next 24 weeks.
Telaprevir (an oral tablet) was given in a single initial dose of 1125 mg, followed by a dose of 750 mg every 8 hours thereafter during the predefined treatment period. Peginterferon alfa-2a was administered at a dose of 180 µg each week, by means of subcutaneous injection. Ribavirin was administered twice daily at a dose of 1000 mg per day for patients weighing less than 75 kg and a dose of 1200 mg per day for patients weighing 75 kg or more.
Randomization was stratified to balance race or ethnic group (black vs. nonblack) and previous virologic response (achievement or nonachievement of undetectable HCV RNA levels) among the four treatment groups. The enrollment was also limited such that neither of the two previous-response strata represented more than 60% of the overall study population. For the first 24 weeks, the treatment assignments were double-blinded for patients in two of the three telaprevir groups (T12PR24 and T24PR48, but not T24P24). At week 24, patients in the T12PR24 group and those in the T24P24 group ended treatment, and treatment continued, through week 48, for patients in the T24PR48 group and those in the PR48 group. Patients randomly assigned to retreatment with peginterferon alfa-2a and ribavirin (the PR48 group) who did not have a sustained virologic response were subsequently offered access to telaprevir, peginterferon alfa-2a, and ribavirin, in a separate study.
Efficacy Assessments
Plasma HCV RNA levels were measured with the use of the COBAS TaqMan HCV assay, version 1.0 (Roche Molecular Systems), with a lower limit of quantification of 30 IU per milliliter and a lower limit of detection of 10 IU per milliliter. Measurements were obtained at various time points during the treatment period, which varied among the assigned treatments: on day 1 and at weeks 1, 2, 3, 4, 8, 12, 16, 20, 24, 32, 36, 40, 44, and 48. All patients with undetectable HCV RNA levels (<10 IU per milliliter) at the end of the treatment period were followed to assess the durability of that response at weeks 4, 12, and 24 after the last dose. In addition, a 48-week assessment was performed in patients in the telaprevir groups who completed the assigned treatment. HCV RNA results were double-blinded through week 24. However, an independent reviewer assessed the HCV RNA levels through week 24 and notified investigators if patients met any of four criteria for discontinuation of all study drugs. These stopping rules were intended to prevent the continuation of treatment in patients who were unlikely to have a response to continued therapy.
The four stopping rules were as follows. The first was a stopping rule for breakthrough from week 4 through week 24, stating that patients must discontinue all study drugs after a confirmed increase in the HCV RNA level of more than 1 log10 unit, as compared with the nadir, during week 4 through week 24 of treatment or an HCV RNA level of more than 100 IU per milliliter in patients who had undetectable HCV RNA levels at a previous time point. The second was a stopping rule for nonresponse at week 4, stating that by week 4 of treatment, patients in the PR48 group must have had a drop of at least 1 log10 unit in the HCV RNA level, as compared with the baseline level, and that patients in the other three groups must have had an HCV RNA level of less than 30 IU per milliliter. The third was a stopping rule for nonresponse at week 12, stating that patients must have had a drop of at least 2 log10 units in the HCV RNA level, as compared with the baseline level, by week 12 of treatment. The fourth was a stopping rule for nonresponse at week 24, stating that patients in the T24PR48 and PR48 groups must have had an undetectable HCV RNA level by week 24 of treatment. All patients who met any of the four stopping rules were considered to have had nonresponse in the final analysis.
Safety Assessments
Safety assessments consisted of physical examinations, recording of adverse events, and serum chemical and hematologic assessments at all study visits, including a safety follow-up visit, 14 days after the last dose of study drugs was administered. Regarding adverse-event reporting, at each visit patients were asked to report any side effects they had had since the previous visit; these were reported on the case report forms. Twelve-lead electrocardiography was performed at the screening visit and 2 weeks after the last dose of medication.
Statistical Analysis
The primary end point was a sustained virologic response, defined as an undetectable plasma HCV RNA level 24 weeks after the last dose of study drugs. The analysis included data for all patients who received at least one dose of study drugs. Assuming a rate of sustained virologic response of 45% in the telaprevir groups and 20% in the control group, we calculated that enrollment of approximately 110 patients in each of the four groups would be required for the study to have a statistical power of 90% to detect a significant difference in rates of sustained virologic response among the groups. Prespecified logistic-regression models were used to evaluate the relationship between response rates and treatment assignment, race or ethnic group, previous virologic response, baseline HCV RNA level, HCV genotype, and age. Three other variables - sex, body-mass index, and presence or absence of cirrhosis - were added post hoc and were also explored.
Results
Study Patients
From February through June 2007, a total of 465 patients were randomly assigned to a treatment group, and 453 received at least one dose of study drugs (Figure 1). The baseline characteristics of the study patients were similar across the four treatment groups, except for a significant difference in the median age (Table 1). The majority of patients (306 of 453 [68%]) were men, and the mean age was 51 years. A total of 89% of the study patients (402 of 453) were white. Most patients (418 of 453 [92%]) had HCV RNA levels of 800,000 IU or more per milliliter at baseline. The majority of patients (265 of 453 [58%]) were infected with HCV genotype 1a. In total, 16% of the study population (74 of 453 patients) had cirrhosis. In addition, 57% of patients (260 of 453) were classified as having had a nonresponse to previous HCV therapy, 36% (162 of 453) as having had a relapse, and 7% (31 of 453) as having had a breakthrough.
Efficacy
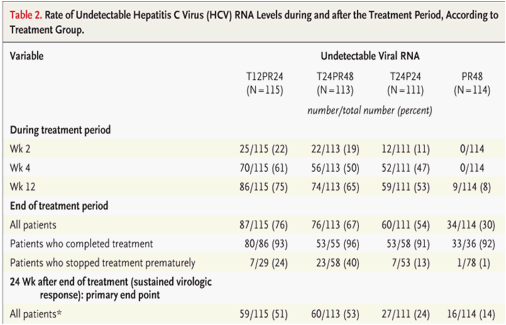
The rate of the primary efficacy end point, a sustained virologic response, was significantly higher in each of the three telaprevir groups - 51% in the T12PR24 group, 53% in the T24PR48 group, and 24% in the T24P24 group - than in the PR48 group (14%; P<0.001, P<0.001, and P=0.02, respectively) (Table 2).
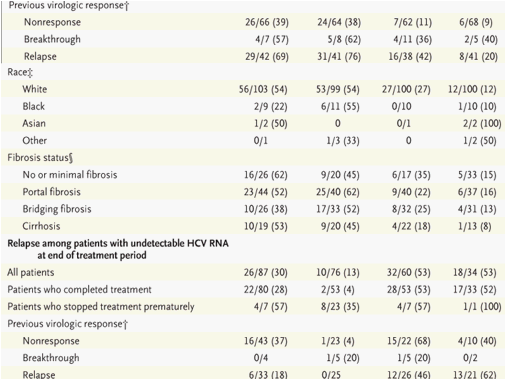
Responses at the end of the treatment period were higher in the telaprevir groups than in the control group, as were the responses at week 4 and week 12 (Table 2). All patients in the telaprevir groups had a virologic response (a drop of at least 2 log10 units in the HCV RNA level, as compared with the baseline level) at week 12. Nine patients in the PR48 group had an early virologic response (at week 12). Rates of sustained virologic response were higher among patients who had previously had a relapse than among those who had not had a response to previous treatment. The rates of sustained virologic response among patients with no previous response were 39% in the T12PR24 group, 38% in the T24PR48 group, and 11% in the T24P24 group, and the rates among patients with a previous relapse were 69% in the T12PR24 group, 76% in the T24PR48 group, and 42% in the T24P24 group. In the PR48 group, the rates were 9% among patients with previous nonresponse and 20% among patients with a previous relapse.
Overall, relapse rates were 30% in the T12PR24 group, 13% in the T24PR48 group, 53% in the T24P24 group, and 53% in the PR48 group. For the patients in the T12PR24 group who completed treatment, relapse rates were 28% overall, 37% among patients with no previous response, and 18% among patients with a previous relapse. For patients in the T24PR48 group who completed treatment, relapse rates were 4% overall, 4% for patients with no previous response, and 0% for patients with a previous relapse.
Rates of viral breakthrough at week 24 were 13% in the T12PR24 group, 12% in the T24PR48 group, 32% in the T24P24 group, and 3% in the PR48 group. In all three telaprevir groups, the majority of patients with viral breakthrough had not had a previous response (Table 3). Most viral breakthroughs were observed during the first 12 weeks of treatment. In the telaprevir groups, the rates of viral breakthrough were higher among patients infected with HCV genotype 1a (24%) than among patients infected with HCV genotype 1b (11%). Most viral breakthroughs were associated with HCV variants with resistance to telaprevir, as previously observed in studies of patients who had not previously been treated.14,15
Logistic-regression analysis showed that a sustained virologic response was significantly associated with assignment to the T12PR24 or T24PR48 group, an undetectable HCV RNA level during a previous period of treatment with peginterferon alfa and ribavirin, and low baseline viral load (<800,000 IU per milliliter).
With the exception of one patient who was lost to follow-up, in all patients who completed a telaprevir regimen and who had a sustained virologic response 24 weeks after the end of the treatment period, the virologic response was maintained at 48 weeks. Seven of 14 patients in the three telaprevir groups who prematurely discontinued treatment and had a sustained virologic response also underwent a follow-up assessment of the viral level at week 48. In these seven patients, the virologic response was maintained 48 weeks after the end of treatment.
Safety
Several of the most common adverse events, such as fatigue and influenza-like symptoms, were consistent with typical interferon-related systemic symptoms (Table 4). However, rash and pruritus were more common in the groups that received telaprevir than in the PR48 group; rash and pruritus were also reported in earlier studies of patients who had not previously been treated.14,15 The overall incidence of any rash-related event was 50% in the T12PR24 group and 60% in the T24PR48 group, as compared with 20% in the PR48 group. The severity of the rash was also greater in the T12PR24 and T24PR48 groups than in the PR48 group.
In general, the rashes in patients in the telaprevir groups were described as maculopapular. The median time to the appearance of a rash of any severity in the telaprevir groups was 7 to 28 days. Severe (grade 3) rash was found in 5% of patients in the T12PR24 group, 4% in the T24PR48 group, 3% in the T24P24 group, and 0% in the control group. Eighteen patients in the telaprevir groups (5%), but none of those in the control group, discontinued treatment because of rash. Rash was monitored during the treatment period; medications including topical antiallergic agents and topical and systemic antipruritic agents were allowed, at the investigator's discretion, for all grades of rash. If discontinuation of study medication was indicated, both ribavirin and telaprevir were stopped while patients continued to receive peginterferon alfa-2a. The median time to treatment discontinuation because of rash in the telaprevir groups was 51 to 64 days (range, 4 to 123) after the start of treatment. Severe rashes resolved in 12 of 14 patients within 9 to 104 days after onset; follow-up data were not available for 2 patients; 6 patients had severe rash necessitating corticosteroid treatment; and 1 was hospitalized for severe rash.
Patients who received telaprevir-based therapy were more likely to discontinue treatment because of an adverse event (50 of 339 [15%]) than were controls, who received peginterferon alfa-2a and ribavirin alone (5 of 114 [4%]). Skin disorders were the most frequent cause of discontinuation among the telaprevir groups (accounting for 22 of the 50 patients who discontinued treatment).
Changes in laboratory values during the study were consistent with those reported in association with the use of peginterferon alfa and ribavirin. A decrease in hemoglobin levels was more common in patients receiving telaprevir-based regimens than in controls (Figure 2) and reversed on discontinuation of telaprevir. Treatment with erythropoietin-stimulating agents was prohibited during the course of the study.
Discussion
The results of this study show a benefit of telaprevir in patients who had not had a sustained virologic response to an initial, full course of peginterferon alfa and ribavirin. As in previous studies in untreated patients, a higher breakthrough rate (32%), higher relapse rate (53%), and lower sustained virologic response rate (24%) were observed in the T24P24 group as compared with the T12PR24 group and the T24PR48 group, again demonstrating the necessity for inclusion of ribavirin in combination with telaprevir and peginterferon alfa.14 Patients' previous response to peginterferon alfa and ribavirin also appeared to influence their response to combined treatment with telaprevir. Those who had previously been documented as having clearing of HCV RNA during therapy and who subsequently had a relapse were the most likely to have a response to telaprevir plus peginterferon alfa-2a and ribavirin (69 to 76% of these patients had a sustained virologic response). Those with a previous nonresponse were less likely to have a response after retreatment, but in this subgroup of patients, the observed response rates of 38% in the T24PR48 group and 39% in the T12PR24 group are the highest reported to date and more than four times the response rate in the control group (9%). Results should be interpreted with caution for patients of African, Hispanic, or Asian ancestry, since only a small number of such patients were included in this study.
Viral breakthrough rates were 12 to 13% in patients receiving telaprevir and peginterferon alfa-2a with ribavirin. Viral breakthrough was more common in the absence of ribavirin, occurred early during therapy, was more common in patients who had not had a response to previous therapy than in patients who had had a relapse after previous therapy, and was more frequently observed in patients infected with HCV genotype 1a, as previously reported in patients who had not been treated.14,15 Among patients who had viral breakthrough during the treatment period, sequencing analysis revealed HCV variants with substitutions at the NS3 protease amino acids 36, 54, 155, and 156, findings that are similar to those in patients who had not previously been treated and who had viral breakthrough while receiving telaprevir. The majority of patients who discontinued therapy because of a stopping rule had the V36M/R155K double variant; all but one of the patients had infection with HCV genotype 1a. Relapse rates were substantially lower when telaprevir was added to the retreatment regimen, as in previous studies.14,15 Relapse occurred less frequently with the T24PR48 regimen than with the T12PR24 regimen. As in other studies of telaprevir, rates of discontinuation due to side effects were higher among patients receiving the three-drug combination, and for those patients, discontinuation was most commonly due to rash.
Rates of sustained virologic response were similar in the T12PR24 and T24PR48 groups, and discontinuation of treatment due to adverse events was less common in the T12PR24 group than in the T24PR48 group. Therefore, the overall efficacy and safety results indicate that the T12PR24 regimen appeared to provide a better riskĐbenefit profile than the T24PR48 regimen. The higher discontinuation rates and the lower relapse rates in the T24PR48 group than in the T12PR24 group suggest that an optimal retreatment regimen may consist of a shorter period of treatment with telaprevir combined with a longer period of treatment with peginterferon alfa and ribavirin. Erythropoietin-stimulating agents were not permitted in this study; however, anemia was more frequently observed in patients who received a telaprevir-based regimen. The study design did not address other possible treatment options, such as 12 weeks of telaprevir plus 48 weeks of peginterferon alfa-2a and ribavirin.
In conclusion, this phase 2 study showed that the addition of telaprevir to current therapy as a retreatment strategy in HCV-infected patients who had not had a sustained response to previous therapy significantly increased the likelihood of eradication of HCV infection.
Supported by Vertex Pharmaceuticals.
|
|
| |
| |
|
|
|