| |
New directly acting antivirals for hepatitis C:
potential for interaction with antiretrovirals
|
| |
| |
Journal of Antimicrobial Chemotherapy Advance Pub March 2010 65(6):1079-1085
"Eventually, HCV treatment may involve a combination of directly acting STAT-C agents, without the requirement for PEG-IFN or ribavirin treatment. In a manner analogous to HIV therapy, this is likely to comprise a combination of protease and polymerase inhibitors,9 which can achieve suppression of viral replication, and be suitably robust against emergence of resistance. In the more immediate future however, single STAT-C agents will be added to PEG-IFN and ribavirin therapy.....Currently, ARV treatment may be adjusted, as far as is practicable, to enable optimal administration of anti-HCV therapy, without compromising ARV efficacy. This will become increasingly complex to manage with the addition of new STAT-C agents. Figure 2 summarizes known an
Conclusions: As more data become available concerning the metabolism, clearance, drug transporter interaction and toxicities of the STAT-C agents in development, potential DDIs with ARVs may be better anticipated. Many interactions are expected between HIV PIs and NNRTIs, and HCV protease inhibitors. It is unlikely that DDIs will preclude treatment of both conditions concurrently; but accurate characterization of these interactions, and prompt recognition of potential dangers by clinicians is important to ensure the safety and efficacy of treatment.
It is also important to note that although DDIs can be theoretically pre-empted and a basis formed for practical management, therapeutic drug monitoring and assessment of individual patient response are important in the management of complicated regimens, particularly when they involve new agents with relatively little experience in the clinical setting."
Kay Seden1,*, David Back2 and Saye Khoo2
1 NIHR Biomedical Research Centre, Royal Liverpool & Broadgreen University Hospitals Trust, Liverpool, UK 2 Department of Pharmacology and Therapeutics, University of Liverpool, Liverpool, UK
* Corresponding author. Department of Pharmacology, University of Liverpool, Pharmacology Research Laboratories, Block H, First Floor, 70 Pembroke Place, Liverpool L69 3GF, UK. Tel/Fax: +44-151-706-2883; E-mail: kseden@liverpool.ac.uk
Abstract
Recent advances in the development of agents that act specifically to inhibit hepatitis C virus (HCV) are set to fundamentally change the way that patients will be treated. New directly acting anti-HCV agents such as protease and polymerase inhibitors will initially be added to standard of care with pegylated interferon-{alpha} and ribavirin. However, future therapy is likely to constitute combinations of agents which act at distinct stages of viral replication and have differing resistance profiles. While directly acting anti-HCV agents will undoubtedly improve treatment outcomes, the introduction of combination therapy may not be without complications in some patient groups. HIV-positive patients who are receiving antiretrovirals (ARVs) are relatively highly represented among those with HCV infection, and are at high risk of drug�drug interactions (DDIs). As combination anti-HCV treatment gradually evolves to resemble anti-HIV therapy, it is essential to consider the increased potential for DDIs in patients receiving combination anti-HCV therapy, and particularly in HCV/HIV-co-infected individuals. Therapeutic drug monitoring is likely to play a role in the clinical management of such interactions.
Introduction
Treatment of hepatitis C virus (HCV) with antivirals aims to achieve a sustained virological response (SVR), which equates to undetectable HCV RNA levels 6 months after completion of therapy. This is associated with reduced progression of liver disease and reduced viral transmission. The standard of care for patients requiring treatment for chronic HCV currently involves a combination of pegylated interferon-{alpha} (PEG-IFN) and ribavirin, with response rates and duration of treatment that vary according to HCV genotype.1,2 Weight-based dosing of ribavirin is associated with better treatment outcomes for certain genotypes.3 Clinical studies have shown that combinations of ribavirin and PEG-IFN can achieve an SVR in only 36%�46% of patients with HCV genotype 1 monoinfection, when treated for 48 weeks.1,4
Due to the limited success rates of current treatment and the well documented adverse event profiles of ribavirin and PEG-IFN, there is evident need for novel, directly acting treatments. Specifically targeted antiviral therapy for HCV (STAT-C) represents a new treatment paradigm with improved patient outcomes. There are several STAT-C agents at various stages of clinical development, including protease inhibitors (PIs)5�8 and nucleoside/non-nucleoside polymerase inhibitors.9,10 Other agents under investigation include novel analogues of ribavirin,11 modified interferons,12 cyclophilin B,13 {alpha} glucosidase inhibitors,14 oligonucleotides15 and immune modulators.16
In patients co-infected with HIV/HCV, progression to cirrhosis, end-stage liver disease and ultimately death is more rapid.17 HCV viral loads may be higher in co-infected patients than those with monoinfection,17 and treatment outcomes in terms of SVR rates may be worse in co-infected patients, particularly with genotype 1 HCV infection.18 No significant difference in efficacy or safety has been reported in co-infected patients treated with PEG-IFN-2a plus ribavirin or PEG-IFN-2b plus ribavirin.19 HCV-associated liver failure is increasing significantly as a cause of death in HIV-positive patients in the post highly active antiretroviral therapy (HAART) era in developed countries.20 It therefore follows that significant changes in HCV therapy could profoundly improve the treatment outcomes of this patient group. However, the addition of combination therapies, which include novel agents, for patients who are taking antiretroviral (ARV) regimens is unlikely to be without complication; clinically significant drug�drug interactions (DDIs) involving ARVs are common, affecting 27% of 159 HIV-infected outpatients in a UK study21 and 23%�26% of 220 HIV-infected outpatients in a study in the Netherlands.22
The future of anti-HCV therapy
Every stage of the HCV life cycle potentially represents a target for STAT-C agents,15 meaning that increasing numbers of novel agents and novel classes of agents are likely to emerge, in a manner similar to ARV therapy. Currently in Phase III trials, the NS3/4A PIs telaprevir (VX-950)5,6 and boceprevir (SCH 503034)8 are likely to be the first STAT-C agents licensed. The development of some of the furthest advanced nucleoside analogue NS5B polymerase inhibitors has been halted due to toxicities; however, R7128, a pro-drug of PSI-6130, is currently in Phase II trials. Figure 1 illustrates the location in the HCV genome of drug targets, including the NS3/4A serine protease and the NS5B RNA-dependent RNA polymerase enzymes, which, being essential for viral replication, are primary targets for anti-HCV therapy.
Figure 1. Important targets for STAT-C therapy in the HCV genome. Derived from information in Pawlotsky et al.15 and Chevaliez and Pawlotsky.46
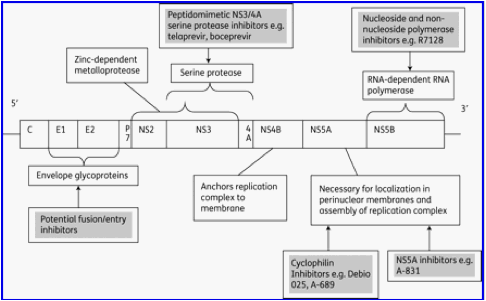
Eventually, HCV treatment may involve a combination of directly acting STAT-C agents, without the requirement for PEG-IFN or ribavirin treatment. In a manner analogous to HIV therapy, this is likely to comprise a combination of protease and polymerase inhibitors,9 which can achieve suppression of viral replication, and be suitably robust against emergence of resistance. In the more immediate future however, single STAT-C agents will be added to PEG-IFN and ribavirin therapy, until there are sufficient effective STAT-C agents licensed with which to afford effective combination therapy with a suitable barrier to resistance. Several studies have confirmed the pivotal role of ribavirin in successful HCV antiviral therapy, despite having little intrinsic antiviral activity. For example, with telaprevir and PEG-IFN, ribavirin was found to increase SVR rates by preventing relapse and emergence of telaprevir resistance.6
To draw another comparison with HIV therapy, it is almost certain that the selection of several agents without overlapping resistance patterns will be required for combination therapy, which will increase both the complexity and risk for DDIs.
ARVs and current anti-HCV treatment: potential for DDIs
As PEG-IFN and ribavirin are expected to remain a fundamental component of anti-HCV treatment in the near future, their potential for interaction with ARVs remains important with the advent of STAT-C agents.
Concomitant administration of abacavir with PEG-IFN and ribavirin has been associated with an increased risk of non-response to anti-HCV therapy,23 and an interaction between abacavir and ribavirin has been suggested. As both drugs are guanosine analogues and have some metabolic pathways in common, an inhibitory competition for phosphorylation is the likely mechanism.24
Combinations of zidovudine with ribavirin and PEG-IFN can lead to increased risk of severe haematological toxicity, including anaemia. The use of zidovudine has been identified as an independent factor contributing to haematological adverse events in patients undergoing ribavirin and PEG-IFN treatment; the combination is not recommended.25
The use of didanosine alongside ribavirin is associated with increased risk of mitochondrial toxicity, which may be attributed to increased exposure to the active metabolite of didanosine, dideoxyadenosine 5'-triphosphate, when didanosine is co-administered with ribavirin.26�28 Toxicity may be severe and co-administration is not recommended. Mitochondrial toxicity has also been observed with combinations of stavudine and ribavirin. In vitro data have shown that ribavirin can inhibit phosphorylation of zidovudine and stavudine. The clinical significance is not clear; however, close monitoring of HIV RNA with this combination is recommended.
In relation to HIV PIs, it has been reported that in HIV/HCV-co-infected patients, serum bilirubin increases following initiation of PEG-IFN and ribavirin were 1.9-fold higher in patients taking an atazanavir-containing regimen.29 Hyperbilirubinaemia is, however, a relatively common side effect of atazanavir treatment, for which there are various risk factors irrespective of anti-HCV treatment.30 Atazanavir inhibits UGT1A1, an enzyme involved in bilirubin conjugation; it is important to note that different genotypes of UGT1A1 (notably *6*6, *7*7 and *6*731) have an impact on enzyme activity, and interindividual variability in the frequency and severity of hyperbilirubinaemia has been observed in patients treated with atazanavir.31
With HIV non-nucleoside reverse transcriptase inhibitors (NNRTIs), if patients receive efavirenz alongside PEG-IFN, monitoring of CNS effects is important, as the incidence of depressive symptoms in patients with HIV/HCV co-infection treated with IFN is reportedly high.32
Currently, ARV treatment may be adjusted, as far as is practicable, to enable optimal administration of anti-HCV therapy, without compromising ARV efficacy. This will become increasingly complex to manage with the addition of new STAT-C agents. Figure 2 summarizes known and potential DDIs between ARVs and anti-HCV drugs in current use and the HCV PIs in late stages of development.
Figure 2. A summary of known and anticipated DDIs between antiretrovirals and anti-HCV drugs in current use and the HCV protease inhibitors in Phase III development.
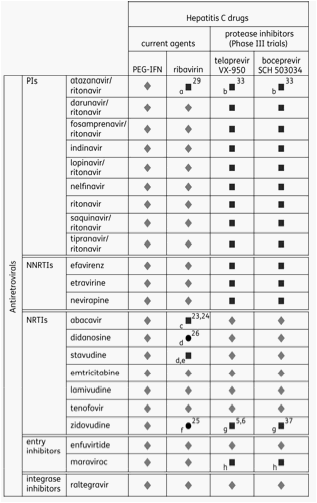

Potential interactions between ARVs and STAT-C agents
As illustrated in Figure 2, few data are currently available to evaluate the potential for DDIs between ARVs and emerging STAT-C agents. Most is known about telaprevir and boceprevir, the HCV PIs which are furthest through the development process.
Although they target serine rather than aspartate proteins, HCV PIs have structural similarities with HIV PIs, and there is evidence to suggest that they share a common route of metabolism.33 In human liver microsomes, the metabolism of telaprevir and boceprevir was substantially inhibited in the presence of relatively low concentrations of ritonavir.33 In addition, on co-dosing either telaprevir or boceprevir with ritonavir in rats, the plasma exposure of both HCV agents was markedly increased. These findings suggest that telaprevir and boceprevir may be primarily or exclusively metabolized by CYP3A. This has implications for co-administration of ARV therapy, as HIV PIs such as lopinavir and darunavir inhibit CYP3A4, whereas the NNRTIs efavirenz, nevirapine and etravirine induce metabolism mediated by this enzyme. Although PIs are able to inhibit CYP3A4, the observed effect may be predominantly due to potent inhibitory action of ritonavir, which is commonly used as a pharmacokinetic enhancer to boost levels of other PIs.
Such interactions, when more comprehensively understood, may not be problematic if appropriately managed. For example, ritonavir could be utilized to dually boost both an HIV PI and an HCV PI. Both ritonavir and newer pharmacokinetic enhancers in development such as GS 935034 could be used to reduce dosing frequency, allowing telaprevir and boceprevir to be dosed less frequently than 8 hourly and thus increasing the likelihood of adherence.33 In addition, there are ongoing studies to develop follow-on compounds of the clinical candidate. For example, systematic structure�activity relationship studies of different regions of boceprevir have shown improved potency and improved pharmacokinetics compared with boceprevir in model systems.35
As a result of structural similarities between some nucleoside reverse transcriptase inhibitors (NRTIs) and HCV NS5B nucleoside polymerase inhibitors, competition for clearance pathways cannot be ruled out. For example, R7128 is a cytidine analogue, structurally related to the HIV NRTI cytidine analogues lamivudine and emtricitabine. Although there may be potential for interaction, relevance to the clinical setting remains to be elucidated.
The non-nucleoside polymerase inhibitor GS 9190 has little potential for inhibition or induction of CYP450 enzymes and is not transported by P-glycoprotein (P-gp) in vitro; therefore, interactions via these mechanisms are unlikely.36
Overlapping toxicity profiles and adverse effects also need to be considered when using STAT-C agents alongside ARV therapy. For example, clinical studies have illustrated that anaemia is a relatively common adverse effect experienced by patients receiving telaprevir5,6 or boceprevir37 therapy. The frequency of anaemia was increased in treatment groups receiving telaprevir alongside PEG-IFN and ribavirin, compared with groups receiving PEG-IFN and ribavirin alone.5,6 This may have implications for patients who are taking zidovudine as part of their ARV regimen, as anaemia is a common adverse reaction with zidovudine treatment. Concomitant use of telaprevir or boceprevir with zidovudine could therefore increase the risk of anaemia, particularly when used in combination with ribavirin, which is likely to be the case. Use of ribavirin with zidovudine is currently not recommended by one of the manufacturers of ribavirin,38 due to anaemia risk.
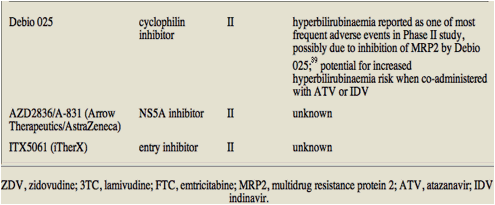
Phase II studies have shown an increased incidence of hyperbilirubinaemia in patients treated with the cyclophilin inhibitor Debio 025 compared with controls.39 This may be due to the inhibition of the multidrug resistance protein 2 (MRP2) transporter by Debio 025,40 resulting in reduced elimination of conjugated bilirubin. As various ARVs are themselves inhibitors of or substrates for MRP2, interactions via this mechanism or UGT1A1, as previously described, cannot be ruled out. It has been suggested that haplotypes of certain genes, such as UGT1A, may be useful predictors of PI-induced hyperbilirubinaemia.41 Genetic diagnostics may be an important application to future HCV, as well as HIV therapies. Interestingly, in the context of co-infection, Debio 025 has also shown inhibitory activity against HIV-1 in vitro.42
Although some Phase II studies of STAT-C agents have reported no significant changes in various laboratory parameters and electrocardiogram readings,5,43 as these new drugs become licensed and clinical experience of their use develops, it is possible that toxicities may emerge, as with ARVs. For example, lipodystrophy with various ARVs,44 mitochondrial toxicity with didanosine28 and, more recently, cardiovascular effects with abacavir.45 It is not inconceivable that STAT-C agents in development could have metabolic or mitochondrial toxicities in common with some ARVs, and future regimens may need to be adapted to minimize risk.
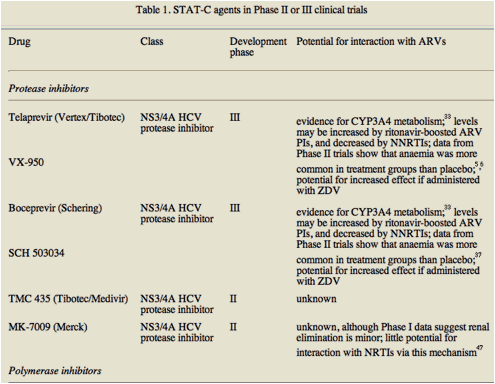
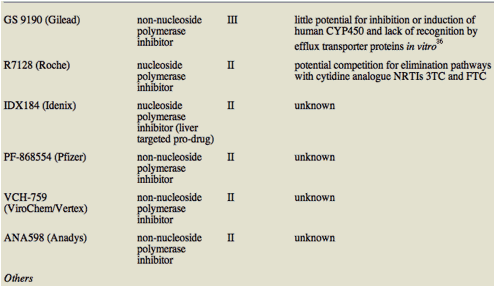
Table 1 lists STAT-C agents in Phase II or III trials and, where data are available, their potential to interact with ARVs.
Conclusions
As more data become available concerning the metabolism, clearance, drug transporter interaction and toxicities of the STAT-C agents in development, potential DDIs with ARVs may be better anticipated. Many interactions are expected between HIV PIs and NNRTIs, and HCV protease inhibitors. It is unlikely that DDIs will preclude treatment of both conditions concurrently; but accurate characterization of these interactions, and prompt recognition of potential dangers by clinicians is important to ensure the safety and efficacy of treatment.
It is also important to note that although DDIs can be theoretically pre-empted and a basis formed for practical management, therapeutic drug monitoring and assessment of individual patient response are important in the management of complicated regimens, particularly when they involve new agents with relatively little experience in the clinical setting.
|
|
| |
| |
|
|
|