 |
|
|
| |
Abacavir/Lamivudine with Fosamprenavir/Ritonavir or Efavirenz in
Underrepresented, Antiretroviral-Naïve, HIV-Infected Subjects (SUPPORT):
Tolerability, Safety, and Efficacy after 24 Weeks
|
| |
| |
Reported by Jules Levin 18th IAC Vienna July 2010
P Kumar1, E DeJesus2, G Huhn3, L Sloan4, F Garcia5, C Small6, H Edelstein7, F Felizarta8,
R Hao9, K Oie10
, L Ross10, B Stancil10, B Ha10, K Pappa10, and the SUPPORT study team
1Georgetown University, Washington DC; 2Orlando Immunology Center, Orlando, FL; 3Ruth M. Rothstein CORE Center, Chicago, IL; 4North Texas
Infectious Disease Consultants, Dallas, TX; 5Valley AIDS Council, Harlingen, TX; 6New York Medical College, Valhalla, NY; 7Alameda County
Medical Center, Oakland, CA; 8Private Practice, Bakersfield, CA; 9Chase Brexton Health Services, Inc., Baltimore, MD; 10GlaxoSmithKline, RTP, NC
From Jules: of interest, after starting HAART in this study CRP levels increased compared to the SHIELD Study where CRP decreased. Its not clear why but when comparing the 2 patient populations, in this study there were much more African-Americans 60% vs 6%, more smokers, and more patients with over 100K viral load and/or <200 CD4s at baseline.


Introduction
The SUPPORT study (COL110408) is a ongoing randomized, 96-week, open-label, prospective, multicenter trial evaluating FPV/r 1400/100 mg once daily (LEXIVA, GlaxoSmithKline, RTP, NC and Norvir, Abbott Laboratories,
North Chicago, IL) versus EFV 600 mg once daily (Sustiva, Bristol-Myers Squibb, Princeton, NJ), both in combination with ABC/3TC 600 mg/300 mg once daily (EPZICOM, GlaxoSmithKline, RTP, NC) in antiretroviral-naïve HLA-B*5701-
negative adults. Results from a planned 24-week interim analysis are presented.
Methods
Subjects were excluded if they had a screening HIV-1 RNA ≤5,000 copies/mL (c/mL), chronic hepatitis B infection (HBsAg+), or if their screening genotype indicated primary resistance mutations to any investigational product. There
were no restrictions on screening CD4 count. Randomization was stratified by screening viral load (
Virologic failure was defined as either virologic non-response (failure to achieve HIV-1 RNA <400 c/mL by week 24 [W24]) or virologic rebound (HIV-1 RNA <400 c/mL by W24 with a subsequent increase to ≥400 c/mL on two consecutive
occasions). In the case of clinically suspected hypersensitivity to ABC, subjects were permitted to substitute a suitable non-abacavir-containing dual nucleoside reverse transcriptase inhibitor (NRTI) backbone and were not discontinued
from the study.
Results
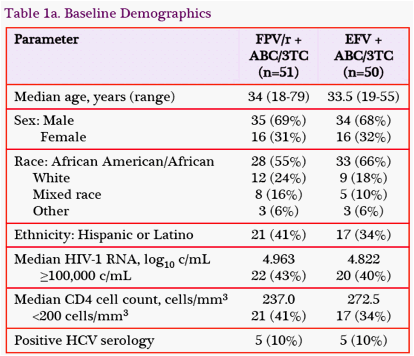
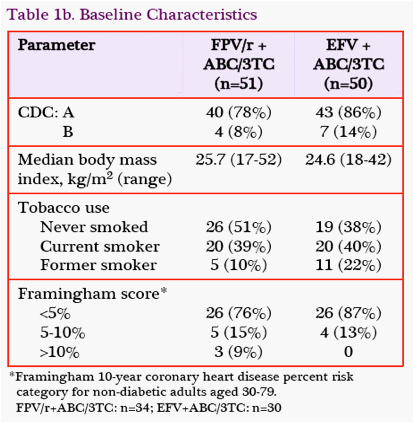
Baseline Demographics and Characteristics: SUPPORT enrolled 101 subjects with a mean age of 34 years (Table 1).
The population was diverse, with 32% women, 60% people of African descent, 13% people of mixed race, and 38% of Hispanic or Latino ethnicity. At baseline, 42% of subjects had HIV-1 RNA ≥100,000 c/mL, and 38% had CD4 cell counts <200 cells/mm3.
Subject Accountability: Most subjects (92%) completed 24 weeks on study. The most common reason for discontinuation was non-compliance (Table 2).
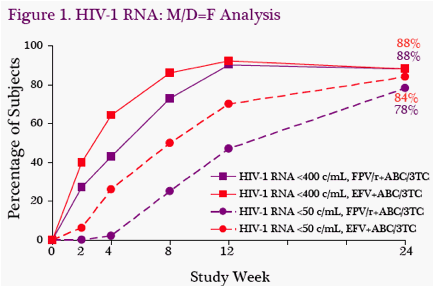
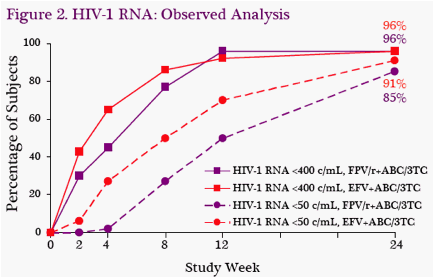
Virologic Supression: Rates of viral suppression were high at W24. Using a missing/discontinuation equals failure analysis, HIV-1 RNA was <50 c/mL in 78%
(40/51) of subjects in FPV/r group and 84% (42/50) in the EFV group (Figure 1). Trends were similar using an observed analysis, in which HIV-1 RNA was <50 c/mL in 85% (40/47) of subjects in FPV/r group and 91% (42/46) in the EFV group (Figure 2).


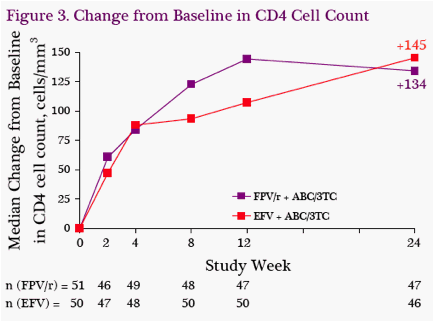
CD4 Cell Count: Changes from baseline to W24 in CD4 cell count were similar, with a median increase of 145 cells/mm3 in the EFV-containing group and 134 cells/mm3 in the FPV/r-containing group (Figure 3).
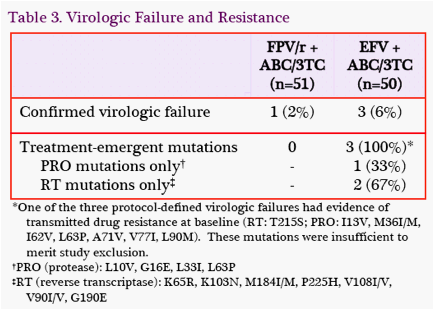
Virologic Failure: There were 4 protocol-defined virologic failures through W24 (Table 3). In the FPV/r-containing group, 1 subject experienced virologic failure and did not have any treatment-emergent mutations. In the EFVcontaining group, 3 subjects experience virologic failure. All 3 had treatment-emergent mutations: 1 with PRO mutations and 2 with RT mutations.
Primary endpoint: The primary endpoint of this study is time to switch of third drug (FPV/r or EFV) or time to development of any treatment-related grade 3 or 4 AE. Through W24, 3 subjects in each group had reached the primary
endpoint (P=0.411). In the FPV/r group, all three patients discontinued study treatment due to AEs: two at 14 and 57 days after study initiation, and the third had a fatal MI 200 days after study initiation. In the EFV group, all three patients
discontinued study treatment due to AEs between 13 and 33 days after study initiation.
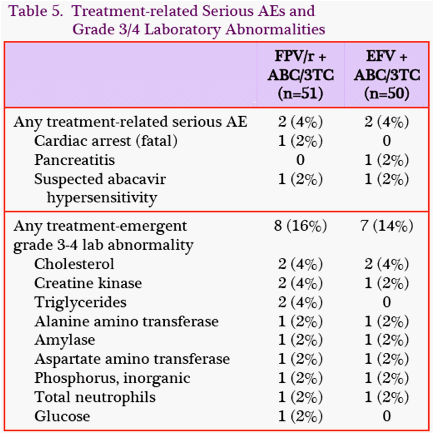
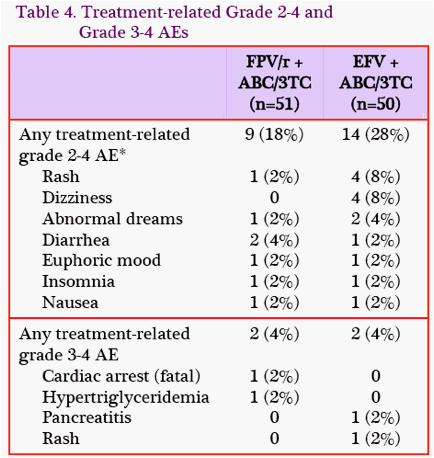
Adverse Events: Through W24, the rate of treatment-related grade 2-4 adverse events (AEs) was lower in the FPV/rcontaining group (9/51, 18%) compared with the EFV-containing group (14/50, 28%) because of EFV-related rash and dizziness (Table 4). Rates of treatment-related grade 3-4 AEs, serious AEs, and grade 3-4 lab abnormalities were similar (Table 5), and 1 subject in each group had a suspected hypersensitivity reaction to ABC.
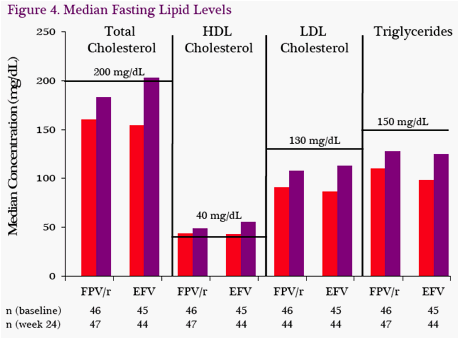
Median Fasting Lipids: At W24, fasting lipid levels increased in both treatment groups (Figure 4). All medians remained below the NCEP guideline cutoffs with the exception of total cholesterol in the EFV group.
Eight FPV/r patients and 3 EFV patients received lipid lowering agents (LLA) during the study. Of these, 6 patients (FPV/r) and 1 patient (EFV) initiated at least one LLA during the study. The fasting lipids analysis was not censored for those patients on LLA.
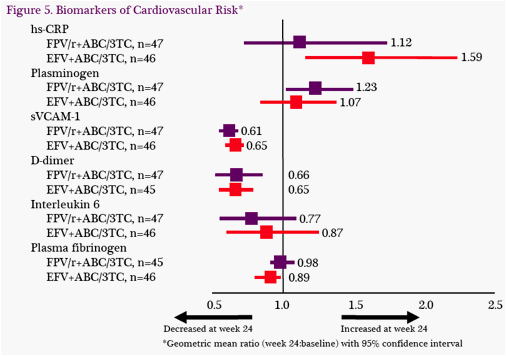
Biomarkers of Cardiovascular Risk: At W24, changes from baseline in inflammatory biomarkers were similar between treatment groups (Figure 5). Hs-CRP and plasminogen tended to increase while all other evaluated markers tended to decrease.
|
| |
|
|
|
|
|