 |
|
|
| |
Comparison of 48 week efficacy and safety of 400mg QD nevirapine (NVP) extended release formulation (Viramune XR) versus 200mg BID nevirapine immediate release formulation (Viramune IR) in combination with emtricitabine/tenofovir in antiretroviral (ARV) naïve HIV-1 infected patients (VERxVE)
|
| |
| |
Reported by Jules Levin
ICAAC Boston Sept 13 2010
J. Gathe1, JR. Bogner2, S. Santiago3, A. Horban4, M. Nelson5,
P. Cahn6, J. Andrade7, D. Spencer8, C. Yong9, T. Nguyen9, W. Zhang9, M. Drulak9 and A. Quinson9*
1.Therapeutic Concepts, Houston TX, USA; 2.Ludwig-Maximillians University, Munich, Germany; 3.Care Resource, Miami FL USA; 4.Warsaw Medical University and Hospital of Infectious Diseases, Warsaw, Poland; 5.Chelsea and
Westminster Hospital, London, UK; 6.Fundacion Huesped, Buenos Aires, Argentina; 7.Antiguo Hospital Civil de Guadalajara, Guadalajara, Mexico; 8.Toga Labs, Edenvale, South Africa; 9.Boehringer-Ingelheim Pharmaceuticals Inc,
Ridgefield CT, USA. *Presenting author
Author Conclusions
This pivotal trial (VERxVE) demonstrated
· Non-inferiority of Viramune XR to Viramune IR
· A similar AE profile for both formulations with numerically fewer AEs for Viramune XR compared with Viramune IR; no new AEs were identified
· Similar efficacy was noted across many PK strata indicating adequate trough drug exposure for Viramune XR
· A similar lipid profile for Viramune XR compared with Viramune IR
· There was consistent relative trough exposure of Viramune XR to Viramune IR across gender, region, and baseline viral-load strata
Once-daily dosing with Viramune XR provides patients with a more convenient treatment regimen with comparable efficacy and safety to Viramune IR
Abstract
Background: Nevirapine (NVP) 400mg once-daily (QD) is the extended release (XR) formulation (Viramune XR) of NVP immediate release (IR) (Viramune IR) 200mg twice-daily (BID). It has the advantage of once-daily (QD) dosing and may offer therapeutic benefit through improved convenience and, consequently, patient compliance. The purpose of this study is to compare the efficacy and safety of Viramune XR with Viramune IR in treatment-naïve patients with HIV-1.
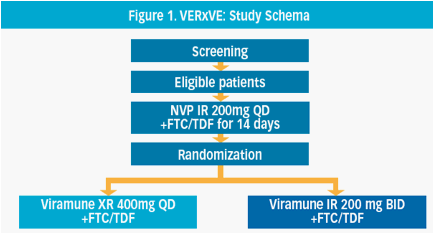
Methods: This double blind, double-dummy parallel group study randomized HIV-1 infected patients >18 years old, viral load (VL) >1,000 copies/mL and CD4+ count < 400 cells/mm3 for males and <250 cells/mm3 for females during screening. After a 14 day lead-in period with 200mg Viramune IR QD, patients were stratified by VL at baseline (≤100,000 or >100,000 copies/mL) and within each stratum randomized 1:1 to Viramune XR or Viramune IR. Primary endpoint was confirmed virologic response (<50 copies/mL) through Week 48 using the time-to-loss of virologic response (TLOVR) algorithm. Cochran's statistic incorporating baseline VL stratum was used to test non-inferiority of Viramune XR to IR using the pre-specified non-inferiority margin of -10%.
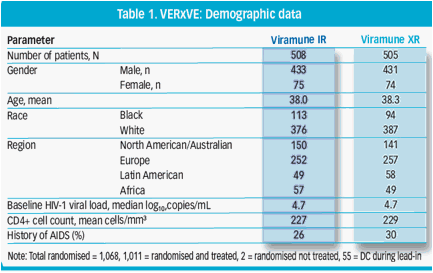
Results: 1,011 patients were randomized and treated. Baseline HIV-1 VL was 4.7 log10 copies/mL for both arms. Demographics, other disease characteristics and extent of exposure to study drug were similar between groups. Virologic response at week 48 was 75.9% (384/506) for IR and 81.0% (409/505) for XR with an adjusted difference of 4.9% in favour of XR and with a 95% CI of (-0.1%, 10.0%), which demonstrated non-inferiority of Viramune XR to IR. This finding was supported by secondary endpoints. The safety profile of XR was similar to IR, but with numerically less adverse events.
Conclusions: Viramune XR was shown to be non-inferior in efficacy to Viramune IR and demonstrated a trend towards improved efficacy and safety with the added convenience of QD dosing. Viramune XR is demonstrated to be an efficacious and well tolerated component of antiretroviral therapy in HIV-1 infected patients.
Introduction
Viramune XR 400mg is the extended release formulation of Viramune IR 200mg twice-daily (BID), which has the advantage of once-daily (QD) dosing. The purpose of this study was to compare the efficacy and safety of Viramune XR with Viramune immediate release (IR) in treatment-naïve patients.
· Viramune XR allows a simple, QD dosing regimen that may increase patient compliance, and subsequently, therapeutic benefit
· In addition, NVP plus emtracitabine/tenofovir (FTC/TDF) recently demonstrated similar efficacy to atazanavir/ritonavir plus TDF/FTC, with a more favourable lipid profile [1]
· This trial compared the efficacy after 48 weeks, and the adverse effect profile up to 48 weeks, of treatment with the XR formulation of 400mg NVP dosed QD versus 200mg BID of the conventional IR formulation, in antiretroviral therapy (ART)-naïve patients with specific CD4+ cell count ranges for males and females as recommended for initiating NVP treatment [2-4]
Methods
Study design
· Multinational, multi-center, randomized, double-blind, double-dummy active controlled trial that evaluated the antiviral efficacy and safety of 400mg QD XR NVP in comparison to 200mg BID IR NVP in combination with emtricitabine/tenofovir (FTC/TDF)
· Participating regions: Americas, Europe, Africa, and Australia
Inclusion criteria
· Patients (≥18 years old), ARV-naïve with viral load (VL)
≥1,000 copies/mL and CD4+count >50-<400 cells/mm3 for males and >50-<250 cells/mm3 for females, at screening, were entered
· Patients with resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs),as wells FTC, TDF, or lamivudine (3TC), were excluded from the study
· Post 14 day lead-in with 200mg IR NVP QD, patients were stratified by VL at baseline (≤100,000 or >100,000 copies/mL) and randomized 1:1 within each stratum to XR NVP 400mg QD (plus placebo) or IR NVP 200mg BID (plus placebo) combined with FTC 200mg plus TDF 300mg, QD, for a minimum 48 weeks (Figure 1)
Primary endpoint
· Sustained virologic response proportion through week 48, defined as two consecutive measurements of VL <50 copies/mL at least two weeks apart (without subsequent viral rebound or change in ARV therapy)
· VL was measured using the Amplicor Ultrasensitive HIV-1 Test, with a lower limit of quantification (LLOQ) of 50 copies/mL
Secondary endpoints
· Time-to-loss of virologic response (TLOVR)
· Time to new AIDS or AIDS-related progression event or death
· Genotypic resistance associated with virologic failure
· Adverse events (including occurrence of rashes and hepatic events)
· Serious adverse events (including AIDS-defining events)
· Laboratory measurement abnormalities
· Occurrence of discontinuations due to adverse events (AEs)
Plasma concentrations of NVP
· NVP trough plasma concentrations were assessed using a validated method of tandem mass spectrometry (LC-MS/MS)
Adverse events
All AEs were recorded, as well as NVP specific AEs, such as rash and hepatic impairment, were collected prospectively
Statistical analysis
· The primary endpoint was non-inferiority of XR NVP to IR NVP; analysis of the primary endpoint used the Amplicor assay profile and time-to-loss of viriologic response (TLOVR) algorithm, as specified by the FDA guidance [5]
· All patients taking at least one dose of study medication were included and weighted treatment difference and corresponding variance calculated based on Cochran's statistic [6] incorporating baseline VL stratum with continuity for variance calculation
· Non-inferiority of XR NVP to IR in terms of efficacy was established if the lower bound of the CI was greater than -10% for the virologic response portion at week 48
Results
Patient disposition
A total of 1,626 patients were enrolled (Figure 2)
· 1,068 received lead-in NVP - the treated set (TS)
· 1,013 patients were randomised
· 1,011 were treated: 505 with XR NVP and 506 with IR NVP - the full analysis set (FAS)

Demographics and HIV baseline characteristics
· Baseline demographic data were well matched between treatment groups (Table 1)
· Extent of exposure to study medication was similar for both treatment group
Efficacy at week 48
Primary endpoint: Non-inferiority of XR NVP to IR NVP
The proportion of patients with sustained virologic response was 75.9% (384/506) for IR and NVP 81.0% (409/505) for XR NVP with an adjusted difference of 4.9% in favour of XR NVP and with a 95% CI of (-0.1%, 10.0%) (Figure 3)
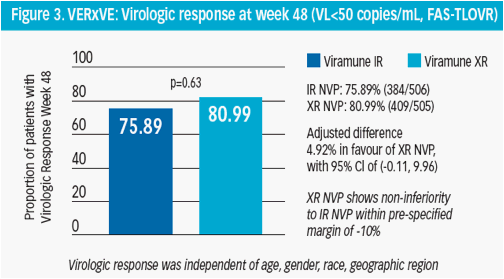
Patients with baseline VL ≤100,000 copies/mL tended to have a higher response proportion than the >100,000 copies/mL
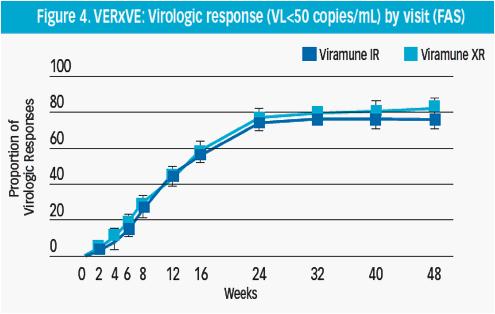
The proportion of virologic responders increased steeply up to 24 weeks of therapy for both the XR and IR NVP treatment groups (Figure 4)
The proportion of XR NVP responders continued to increase from week 24, peaking at week 48
No correlation was observed between the primary endpoint and age, gender, race or geographical location
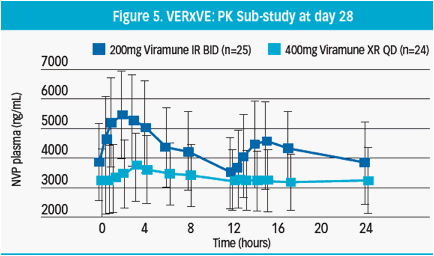
Pharmacokinetic sub-study Day 28
Approximately 30 patients from each treatment arm took part in a pharmacokinetic sub-study on day 28 (visit 4)
· Hospitalised patients gave blood samples intensively for 24 hrs, following morning NVP administration
· Plasma NVP levels were measured by tandem mass spectrometry (LC-MS/MS) and mean (±SD) concentrations determined for BID and QD dosing groups
Mean (±SD) NVP plasma concentrations for the 24-hour period on day 28 were measured (Figure 5)
· Plasma levels from XR NVP 400mg QD were lower, but with less fluctuation, than those provided by IR NVP 200mg BID throughout
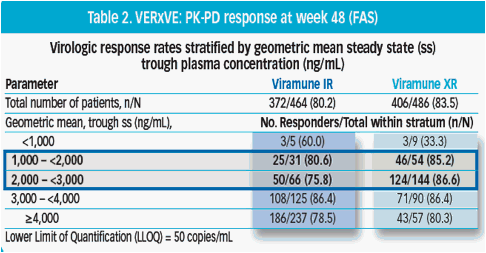
Pharmacokinetic-pharmacodynamic response at week 48
Trough level of NVP concentration appeared to have no effect on virologic response, down to a minimal level of 1,000ng/mL (Table 2)
· Patients in the 1,000 - 2,000ng/mL trough group exhibited comparable virologic response to those in the ≥2,000 ng/ml groups

Changes in lipid profile during IR NVP and XR NVP treatment
· Increases were noted for total cholesterol, LDL-c, and HDL-c, for both formulations. Of note, the total cholesterol/HDL-c ratio decreased with both formulations (Table 3)
· A median decrease was noted in the level of triglycerides for both formulations
Adverse events
A summary of adverse events for the randomised phase is shown in Table 4:
· Similar rates of overall AEs, and AEs leading to discontinuation, were observed in both XR NVP and IR NVP treatment groups
· There were five deaths in the IR NVP group and one in the XR NVP group
- Five deaths occurred during the treatment phase: two due to cardiovascular events and two had infectious disease and one with events in both these classes
- One patient died due to respiratory alkalosis after study medication was stopped
-- None of these fatalities were related to study medication as judged by the investigators

References
1. Soriano V, Arasteh K, Migrone H, Lutz T, Opravil M, Andrade-Villanueva J, Antunes F, i Di Perri G, Podzamczer D, Taylor S, Gellermann HJ and de Rossi L, on behalf of the ARTEN investigators. Week 48 ARTEN results. 2010 (Submitted)
2. Gazzard BG, Anderson J, Babiker A, et al. HIV Med 2008;9:563-608
3. EACS Guidelines: Clinical management and treatment of HIV infected adults in Europe (2009).
http://www.europeanaidsclinicalsociety.org/guidelinespdf/1_Treatment_of_HIV_Infected_Adults.pdf Accessed
April 2010
4. Panel on antiretroviral guidelines for adults and adolescents. Department of Health and Human Services Dec 1, 2009, 1-16. Available at http://aidsinfo.nih.gov/contentfiles/AdultandAdolescentGL.pdf Accessed April 27, 2010
5. Guidance for industry: antiretroviral drugs using plasma HIV RNA measurements - clinical considerations for accelerated and traditional approval. US department of Health and Human Services, Food and Drug Administration,
Center for Drug Evaluation and Research (CDER) 2002: 1-21
6. Cochran W. Biometrics 1954;10:417-451
|
| |
|
|
|
|
|