 |
|
|
| |
11th International Workshop on Clinical Pharmacology
of HIV Therapy (April 2010) Report
|
| |
| |
Courtney V. Fletcher, Pharm.D.
Dean and Professor
College of Pharmacy
University of Nebraska Medical Center
Abbreviations
%CV, percent coefficient of variation
ABC, abacavir
ACTG, adult AIDS clinical trials group
APV, amprenavir
ARV, antiretroviral drug
ART, antiretroviral drug therapy
AUC, area under the concentration-time curve
ATV, atazanavir
Cmin, minimum drug concentration
CNS, central nervous system
CSF, cerebrospinal fluid
CYP, cytochrome P450 drug metabolizing enzymes
DRV, darunavir
ddI, didanosine
EFV, efavirenz
EVG, elvitegravir
FTC, emtricitabine
ETR, etravirine
fAPV, fosamprenavir
IC50, concentration of drug required to inhibit viral replication in vitro by 50%
IDV, indinavir
IM, intramuscular
IQ, inhibitory quotient
3TC, lamivudine
LPV, lopinavir
MVC, maraviroc
NVP, nevirapine
NRTI, nucleoside reverse transcriptase inhibitor
NNRTI, non-nucleoside reverse transcriptase inhibitor
PACTG, pediatric AIDS clinical trials group
PBMCs, peripheral blood mononuclear cells
PD, pharmacodynamic
PG, pharmacogenetics/pharmacogenomics
PK, pharmacokinetic
PI, inhibitor of HIV protease
r or RTV, ritonavir
RAL, raltegravir
SQV, saquinavir
SC, subcutaneous
TDM, therapeutic drug monitoring
TDF, tenofovir disoproxil fumarate
TFV, tenofovir
TPV, tipranavir
ZDV, zidovudine
The 11th International Workshop on Clinical Pharmacology of HIV Therapy was held April 7-9, 2008 in Sorrento, Italy. This year's workshop featured 67 abstracts, five invited lectures, two round table discussions and a clinical case discussion session. Dr. Giovanni DiPerri, M.D., opened the conference with a presentation on the evolution of the HIV epidemic in Italy. Dr. Ed Acosta from the University of Alabama at Birmingham discussed the pharmacology and development of antiviral agents for influenza and potential for drug interactions with ARVs, and Dr. Helen McIlleron from the University of Cape Town discussed the pharmacologic challenges of treatment of HIV-infection and tuberculosis.
Dr. Christian Funck-Brentano gave an invited lecture on the pharmacology and genetics of QTc prolongation. This was a very timely lecture given the FDA's request that all manufacturers of protease inhibitors conduct a QT study to evaluate the effect these drugs have on QT and PR intervals. A prolonged QT interval is concerning because it can increase the risk for serious abnormal heart rhythms; the most serious and life-threatening is known as torsades de pointes. The PR interval reflects the electrical signals in the heart responsible for generating a heartbeat. A drug-induced prolonged PT interval can cause the heartbeat to slow and potentially stop. He noted that risk for torsades is often a cumulative risk starting with host genetic susceptibility (e.g. congenital long QT syndrome) that is then exaggerated by underlying cardiac disease, electrolyte abnormalities, drugs that may prolong QT, drugs that may prolong QT and that can affect metabolism of such drugs (e.g. potentially ritonavir), and the administration of multiple drugs that may prolong the QT. Among data he presented was a prospective cross-sectional study of 978 HIV-infected patients who had a digital electrocardiographic and measured QT interval. This study found that PIs (LPV/r and ATV/r), as well as NNRTIs and NRTIs, were not independently associated with QTc prolongation (Br J Clin Pharmacol 2009;67:76-82). Interestingly, the FDA has recently sent (February 23, 2010) an email warning of preliminary data from a QT-PR study of SQV/RTV (1000/100 mg) given to healthy individuals conducted by Roche. While the FDA noted their analysis was still ongoing, they did state there was a dose-dependent prolongation of the QT and PR intervals, and that patients taking other drugs established to cause QT interval prolongation (such as antiarrhythmics) or persons known to have a prolonged QT interval may be at an increased risk for QT prolongation if also taking SQV/RTV. The determination of whether SQV/RTV has a true independent risk for QT prolongation needs to await the final analysis of this study. However, the latter warning from the FDA of caution if prescribing these drugs in persons who already have some increased risk for QT prolongation is consistent with the message of Dr. Funck-Brentano, and clinicians are advised to exercise caution with these drug combinations in this setting.
The International Workshops on Clinical Pharmacology of HIV Therapy have always provided a venue for interactions of scientists from academia, industry and regulatory authorities (e.g. FDA). This is one of the clear strengths and almost unique characteristics of this Workshop. At the 11th Workshop, Dr. Sarah Robertson with the FDA presented a lecture on the incorporation of pharmacogenetic (PG) information into drug development and drug labeling, which was followed by a round table discussion. The issues discussed can be illustrated with the present situation regarding use of abacavir (ABC) and HLA-B*5701 testing for an increased risk of hypersensitivity reaction (HSR). The FDA-approved label for ABC recommends screening for HLA-B*5701 prior to initiating therapy with ABC. In the PREDICT study, such screening eliminated immunologic ABC HSR compared with standard of care (N Engl J Med, 2008. 358:568-79). My view, and I believe that of most experts in the field, is that HLA-B*5701 screening prior to initiating ABC therapy is the standard of care. Thus the conundrum of whether the FDA-approved label adequately reflects the strength of the PG data for ABC and should HLA-B*5701 screening be recommended or required. These issues extend to several other drugs (e.g. Plavix and screening for poor CYP2C19 metabolism) and will increase as the PG field moves forward. It was a lively, important and timely debate. This meeting is one of the very few venues where this kind of interchange takes place, and I think this is one of the most important characteristics of this meeting.
The abstracts that I have chosen to review in this report are those I felt were well designed, provocative, and had important clinical implications or relevance. I will discuss these abstracts in two sections: Drug-Drug Interactions and Pharmacokinetic-Pharmacodynamic-Pharmacogenetic (PK-PD-PG) Characteristics of ARVs and Therapeutic Implications.
I. Drug-Drug Interactions
The PK of adjusted doses of LPV/r in South African HIV-infected persons were presented in Abstract 47. In this study, patients virologically suppressed on a LPV/r-containing regimen (400/100 mg twice daily) were started on rifampin (RIF) 600 mg/day. The LPV/r dose was increased to 600/150 mg twice daily after 7 days, and increased again to 800/200 mg twice daily after another week. The table below gives the LPV trough concentrations in the 21 persons who participated.

The authors reported that treatment was generally well tolerated with 2 persons developing grade 3/4 aminotransferase elevation. Past studies in healthy volunteers of increased doses of LPV/r when given with RIF have been quite poorly tolerated. These data are encouraging the double-dose of LPV/r overcomes much of the potent CYP enzyme inducing effect, RIF and this strategy is associated with less hepatotoxicity than seen in healthy volunteers. However, note the difference in the lower end of the interquartile ranges (IQR) of the baseline (before RIF) and the 800/200 twice-daily regimen. With the dose of 800/200 twice daily, approximately 25% of the patients still had LPV trough concentrations less than 1 mg/L, the commonly accepted minimally effective concentration. This is a substantially higher proportion than pre-RIF and in my opinion, an unacceptably high proportion. Thus, additional work needs to be done on dose finding, and clearly, it will take a clinical trial to show that this strategy is safe and effective.
The effect of etravirine (ETR) alone and with boosted PIs on the PK of the investigational integrase inhibitor, GSK1349572 (hereafter referred to as GSK572) were presented in Abstract 26. In this study of 16 healthy volunteers, coadministration of ETR and GSK572 significantly decreased GSK572 concentrations: the mean ratio of the GSK572 AUC when given with ETV vs. without was 0.29, indicating a 70% decrease. When ETR and either LPV/r or DRV/r were given with GSK572 the inductive effect of ETR was largely overcome. The AUC of GSK572 was not reduced (though no ratio was actually reported) when ETR was given with LPV/r, and with DRV/r the AUC of GSK572 was reduced 25%. These data from healthy volunteers suggest that GSK572 cannot be given with ETR, but might be able to be coadministered if ETR is combined with LPV/r or DRV/r.
The pharmacokinetics of DRV were evaluated when combined with RTV or the investigational CYP inhibitor GS9350 (Abstract 28). In this study in 33 healthy volunteers, participants received DRV/RTV at 800/100 mg once daily or DRV/GS9350 at 800/150 mg once daily. The geometric mean ratios (and 90% confidence interval) of the PK parameters for DRV/GS9350 vs. DRV/RTV are shown the table below. I have also included the results of a similar study conducted with ATV given either as ATV/RTV (300/100 once daily) or ATV 300 mg given either with 100 or 150 mg of GS9350 once daily. This ATV study was reported at ICAAC in 2009 (Abstract A1-1301). Only the PK values were given for ATV combined with 150 mg of GS9350, presumably because the 100 mg dose of GS9350 did not achieve bioequivalence. Combined, these abstracts give a more complete picture of the boosting effect of GS9350.
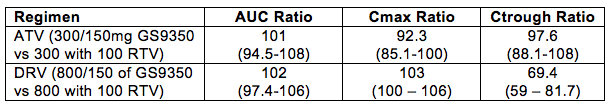
The data from this table show that for ATV, the AUC, Cmax and Ctrough when given with 150 mg of GS9350 are bioequivalent with 100 mg of RTV. For DRV, the AUC and Cmax are bioequivalent also when given with 150 mg of GS9350 compared with 100 mg of RTV. The DRV Ctrough was not bioequivalent, and this arose because of an increase in DRV concentrations at the trough time point. This increase is not typical for DRV, did not occur in the study with ATV, and thus I think is just one of those unusual and unexplainable things that can happen in clinical studies. Collectively, these data to me provide strong support the boosting effect of 150 mg of GS9350 is equivalent to 100 mg of RTV when given with either 300 mg of ATV or 800 mg of DRV.
I want to make a brief mention of Abstract 31, which reported on patterns and use of complementary and alternative medicines (CAM) among 1000 HIV-infected persons attending one clinic in Spain. Overall, 584 (55.5%) of these patients reported using some CAM products within the previous year. Concerns for potential drug-CAM interactions were identified in 36% of the CAM users. The most important message here is for clinicians to understand the frequent use of CAM products and the potential for interactions, and to ask the persons they take care of if they are using any of these products - - and for patients to tell their providers about any of these products they are taking. Knowledge of what is being taken is an essential prerequisite to understanding the potential for any adverse drug-CAM interactions.
II. PK-PD-PG Characteristics of ARVs and Therapeutic Implications
Abstracts 3 and 38 illustrate one challenge of drug development for the treatment of HIV infection, which is to find suitable doses of ARV agents for the newborn to the older (to geriatric) individual. In these abstracts the PK of TDF were evaluated in HIV-infected pregnant women and their newborns (HPTN 057, Abstract 3) and in older adults (Abstract 38, median age 43 years). In the HPTN study, a single dose of 900 mg of TDF was given to women at the onset of labor or 4 hours prior to a C-section, and newborns received a 6 mg/kg dose at birth and 72 and 120 hours after birth. 36 mother-infant pairs were evaluated. The median maternal TFV concentration at delivery was 200 ng/mL and the cord blood concentration was 123 ng/mL, indicating a placental passage penetration ratio of 0.6. A target of 50 ng/mL was established in this study and 84% of the cord blood samples had TFV concentrations above this threshold. This suggests to me that the maternal dose of 900 mg may be the appropriate dose. However, at the time of the first dose to the newborns, predose concentrations were greater than 50 ng/mL in only 22%. Predose concentrations before the 2nd and 3rd doses following birth were also less than 50 ng/mL, indicating the TDF dosing regimen in the newborns was not maintaining concentrations above the 50 ng/mL threshold. The investigators stated they planned to open a 4th cohort that would evaluate daily dosing to the newborns. The need for a 4th cohort speaks loudly as to the challenges in finding the right dose for pregnant women and for newborns. I applaud these investigators for their persistence to get the TDF dose right.
At somewhat of the other end of the age spectrum, 1031 adults attending the Johns Hopkins HIV clinic and receiving TDF were evaluated to assess the impact of age on renal function. These investigators found that TDF use was associated with a loss of renal function, as assessed by estimated glomerular filtration rate (eGFR), and the loss was a function of the duration of TDF use. Age was independently associated with a loss of renal function with older age associated with a greater loss of renal function, and a greater adverse effect of TDF on renal function. The HIV-infected population is aging due to the success of ARV therapy, and approximately 30% of new cases of HIV infection in the US are more than 45 years. We increasingly will need to understand the PK of ARVs in the older adult and the adverse effects, if any, of the ARVs we use on organ function.
Abstract 17 was an interesting comparison of the intracellular concentrations of ATV when given with or without RTV. 29 patients were included in the study with 14 receiving unboosted ATV and 15 receiving ATV and RTV. Not surprising was the finding that intracellular concentrations were higher when ATV was given with RTV than when it was given unboosted (median intracellular troughs of 1032 ng/mL vs 328 ng/mL). What was interesting was that whether ATV was given with RTV or not, intracellular concentrations were higher than plasma concentrations. For example, in the boosted ATV group, the median plasma trough concentration was 543 ng/mL, and the median intracellular concentration was 1032 ng/mL. The cellular penetration ratios for unboosted and boosted ATV were comparable at 1.9 and 2, respectively. These data therefore indicate that ATV accumulates in PBMCs, which suggests a role of transporters in regulation of intracellular concentrations. It will be interesting to obtain similar data for other PIs, and to see if the comparative data help explain the potency and durability of PI regimens.
Two contemporary issues in ARV therapeutics are the need for comparative PK data in HIV-infected persons of generic with brand agents, and whether lower dose regimens might be as effective as the standard dose. These issues were evaluated in Abstract 35. 20 HIV-infected persons participated in this study. 10 received the brand pediatric tablet of LPV/r, 200/50 mg BID for 2 weeks, and 10 received a generic (GPO, Thailand) LPV/r tablet at the same dose. After 2 weeks, the participants crossed over to receive the alternative formulation. Pharmacokinetic evaluations demonstrated the generic formulation met the FDA definition of bioequivalence for LPV and RTV. However, in these Thai subjects, who had a median body weight of 59.8 kg, the reduced LPV/r dose of 200/50 mg BID did not achieve minimum threshold concentrations in 10 (50%) subjects. Of these 10 participants, four were receiving the generic formulation and six the brand formulation. These data, in this small body weight population, are pretty compelling to me that use of a reduced LPV/r dose is likely to place subjects at a higher risk of virologic failure. I don't see any rational argument for further studies of this low dose strategy with LPV/r.
Scientists at GSK stirred up a bit of controversy with their evaluation of the PK/PD properties of their investigational integrase inhibitor, GSK572, along with raltegravir (RAL) and elvitegravir (EVG). They used PK parameters and viral load reductions obtained for these drugs from clinical trials, and calculated inhibitory quotients (IQs) using protein-binding adjusted IC50 values, in a PK/PD evaluation of all drugs combined. They found when the PK parameters were adjusted for IQ, that all of these integrase inhibitors showed a very similar concentration-response profile, including RAL. Of the PK parameters, the antiviral effect was primarily driven by the trough concentration and the trough-adjusted IQ was the best predictor of anti-HIV activity. What is interesting here is that these data suggest that the integrase inhibitors are more similar than different with respect to their concentration-response relationships. This suggestion is consistent with the individual data for GSK572 and EVG, but is inconsistent with the available data for RAL, where no clear concentration-response relationship has been shown. Some pharmacologists have argued that the lack of such a relationship for RAL is simply because the dose range studied is so small, and the interpatient variability so large, that the relationship has been obscured. The data from this study (Abstract 52) would support that argument. I don't think this changes anything for RAL. The safety and efficacy of the drug have clearly been demonstrated in clinical trials at the 400 mg twice-daily dose. However, it does indicate there is some minimally effective threshold concentration, and not just any dose will do and that some drug-drug interactions (e.g. with rifampin) may well be clinically significant. Additionally, these data would suggest that new integrase inhibitors would benefit from broader dose ranging studies, as has been done for EVG and GSK572, so that some knowledge of where that breakpoint is can be learned early in development.
All in all, an outstanding conference that moves the field of HIV therapeutics forward by providing a venue for the presentation and discussion of pharmacologic studies of ARV agents. I am looking forward to the 2011 conference, which will be in the USA.
|
| |
|
|
|
|
|