 |
|
|
| |
HCV Drug Resistance
|
| |
| |
from Jules: This is a controversial issue & was at this just completed EASL. Where a number of researchers said flatly 'we will be able to re-use HCV protease inhibitors after patients develop resistance if we wait 2 years for the mutations to disappear & this was pretty much said with regards to telaprevir following the oral presentation by Sullivan of Vertex reporting that mutations mostly disappear after 1.5 years following stopping telaprevir therapy'. This may end up being true but right now this is just a theory without any evidence, no data to support this supposition, it cannot be considered true until a study or real clinical evidence supports this. Until we have data on this question one cannot say flatly patients will be able to re-use a HCV protease after resistance and receive viral load reductions from the drug. Will such a study ever be done, I don't know. There are many other classes of HCV drugs in development so in the future patients ought to be able to switch to a regimen of 2-4 oral drugs with or without peg/rbv without a protease, so the question may be moot. Still I think we need to clarify this. One issue is that the Sullivan study did not use ultra-deep sequencing, which in this study below shows mutations missed by clonal sequencing are picked up by UDS. At baseline before therapy mutations pre-exist, so 2 years later you would expect these same pre-existing mutations to be present but HCV multi-drug therapy suppresses these mutations and SVR still has been achieved evidenced by the studies with telaprevir & boceprevir, but the question is will these mutations be further enriched after failing therapy, will mutations accumulate to a significant degree if a patient remains on failing therapy, will compensatory mutations accumulate, and will these situations persist & affect re-treatment with a PI, I don't know the answer to these questions and I don't think anyone does. Perhaps the prediction that this will not be a problem is correct but we don't know & we have not studied this adequately.
AASLD: Ultra-Deep Sequencing of the NS3 and NS5B Regions Detects Pre-Existing Resistant Variants to Direct Acting Antivirals (DAA) in HCV Genotype 1 Treatment-Naïve Infected Patients - (11/29/10)
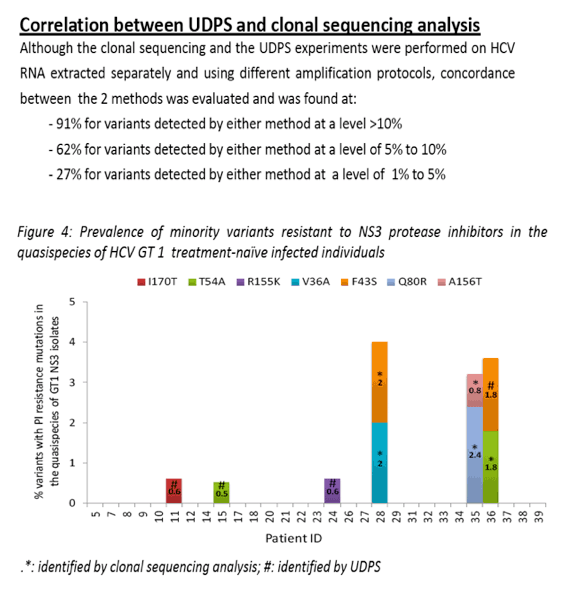
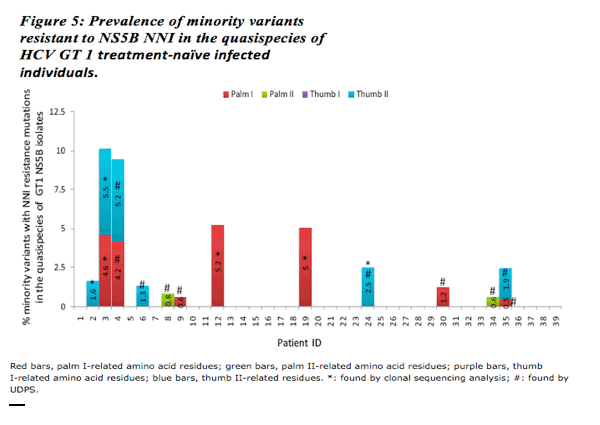
"Minority variants resistant to PI or NNI were detected in 3 and 10 subjects respectively at frequencies that ranged from 0.6 to 14%. Concordance between UDPS and molecular clonal sequencing was 87% for MVs detected (by either method) at a level >10%; 68% for MVs detected at a level of 5% to 10%; and 26% for MVs detected at a level of 1% to 5%."
ULTRA-DEEP SEQUENCING OF THE NS3 AND NS5B REGIONS DETECTS PRE-EXISTING RESISTANT VARI- ANTS TO DIRECT ACTING ANTIVIRALS (DAA) IN HCV GENOTYPE 1 INFECTED PATIENTS NAïVE
AASLD ABSTRACT 2010 October
Severine Margeridon4, Sophie Le Pogam2, Tommy F. Liu4, Bozena Hanczaruk3, Todd E. Arnold3, Birgitte B. Simen3, Nancy Shulman1, Robert W. Shafer4, Isabel Najera2; 1Virology - Pharma Research Early Development, Roche Pharmaceuticals, Palo Alto, CA; 2Virol- ogy - Discovery, Roche Pharmaceuticals, Palo Alto, CA; 3454 Life- sciences, a Roche Company, Branford, CT; 4Infectious Diseases and Geographic Medicine, Stanford University Medical School, Stanford, CA
Background: Naturally occurring HCV drug-resistance minority variants (MVs) that are not detectable by standard consensus sequencing may have an impact on the effectiveness of HCV inhibitors. We used ultradeep pyrosequencing (UDPS) and molecular clonal sequencing to assess the prevalence of HCV drug-resistant minority variants to protease (PI), nucleoside ana- log (NA), and nonnucleoside polymerase inhibitors (NNI) in serum samples from DAA-naïve HCV infected subjects.
Methods: Samples were obtained from 39 DAA treatment naive HCV-infected subjects (27 genotype 1a and 12 genotype 1b). Molecular clonal sequencing and UDPS were performed span- ning those regions of the NS3 protease,and of the NS5B poly- merase including amino acid residues known to confer resistance to PIs, NAs and NNIs. The median viral load was 6.6 log10 IU/ml (range 5.1 - 7.6). UDPS was performed using the GS FLX Titanium platform (454 / Roche Life Sciences) at a median coverage per base of ~3,500. MVs for the UDPS were defined as mutations present in ≥0.5% of UDPS reads..whereas for the clonal sequencing analysis, MVs were defined as muta- tions present in ≥1 molecular clone.
Results: Plasmid control experiments identified a UDPS error rate of 0.1% - resulting from a combination of PCR and pyrosequencing error. No NA resistance mutation was detected by UDPS nor through molecular clonal sequencing. Minority variants resistant to PI or NNI were detected in 3 and 10 subjects respectively at frequencies that ranged from 0.6 to 14%. Concordance between UDPS and molecular clonal sequencing was 87% for MVs detected (by either method) at a level >10%; 68% for MVs detected at a level of 5% to 10%; and 26% for MVs detected at a level of 1% to 5%.
Conclusions: UDPS is a powerful technique for detecting minority variants within viral quasispecies. Optimization of a number of factors such as amplification error rate, input viral load, and the refining the sensitivity of the technique is ongoing. Comparison of results from molecular cloning and UDPS showed that no nucleoside analogs DRM as detected through either technique. Both techniques, however, detected the most prevalent NNI-DRMs with relative high concordance for MVs present at levels > 5%.
|
| |
|
|
|
|
|