| |
Patient acceptance of universal screening for hepatitis C virus infectionAntiviral activity of the hepatitis C virus polymerase inhibitor filibuvir in genotype 1-infected patients
|
| |
| |
Download the PDF here
HEPATOLOGY, July 2011
Frank Wagner,1 Robert Thompson,2 Constantino Kantaridis,3 Paul Simpson,4 Philip J. F. Troke,4 Shyla Jagannatha,5 Srividya Neelakantan,5 Vivek S. Purohit,5 and Jennifer L. Hammond
From the 1Charite Research Organisation, Charite Universita tsmedizin Berlin, Berlin, Germany; 2University of Florida, Center for Clinical Trials Research, FL; 3Pfizer Clinical Research Unit, Pfizer, Brussels, Belgium; 4Pfizer Global Research, Sandwich, Kent, UK; and 5Pfizer Worldwide Biopharmaceuticals, New London, CT.
"This is the first report of the antiviral activity and safety of filibuvir in HCV-infected patients. Data from these two phase 1b studies showed that filibuvir potently inhibited viral replication in a dose-dependent manner in patients infected with HCV genotype 1. The mean maximum reduction in HCV RNA concentrations ranged from -0.97 log10 IU/mL (100 mg BID) to -2.30 log10 IU/mL (700 mg BID). In study 1, the mean maximum reductions in HCV RNA were statistically greater than placebo for all filibuvir doses evaluated."
ABSTRACT
More effective and better-tolerated therapies are needed for chronic hepatitis C virus (HCV) infection. Among the direct-acting anti-HCV agents in development is the nonstructural 5B protein (NS5B polymerase) non-nucleoside inhibitor filibuvir. We investigated the antiviral activity, pharmacokinetics, safety, and tolerability of multiple doses of filibuvir in treatment-naive and treatment-experienced patients who were chronically infected with HCV genotype 1 in two phase 1b clinical studies (study 1 was a randomized, placebo-controlled dose escalation study and study 2 was a nonrandomized, open-label study). The filibuvir doses evaluated ranged from 200-1400 mg daily, and the duration of dosing ranged from 3-10 days. Genotypic changes in the NS5B nucleotide sequence following short-term filibuvir therapy were also assessed. Filibuvir potently inhibited viral replication in a dose-dependent manner. Mean maximum HCV RNA change from baseline ranged from -0.97 log10 IU/mL with filibuvir given at 100 mg twice daily to -2.30 log10 IU/mL with filibuvir given at 700 mg twice daily in treatment-naive patients. In treatment-experienced patients, an HCV RNA reduction of 2.20 log10 IU/mL was achieved with filibuvir given at 450 mg twice daily. Filibuvir was well tolerated in both studies. Adverse events were mild or moderate in severity. No discontinuations, serious adverse events, or deaths were reported. NS5B sequencing identified residue 423 as the predominant site of mutation after filibuvir dosing. Conclusion: Filibuvir administration resulted in significant reductions in HCV RNA concentrations at doses that were well tolerated in patients infected with HCV genotype 1. Filibuvir is currently being evaluated in combination with pegylated interferon alfa 2a plus ribavirin in treatment-naive patients. (Hepatology 2011;)
Hepatitis C virus (HCV) infection affects approximately 180 million people worldwide1 and is a leading cause of chronic liver disease.2 The current standard of care for chronic HCV infection is a combination of pegylated interferon alfa (pegIFN) and ribavirin (RBV).3 In treatment-naive patients who are infected with HCV genotype 1, administration of pegIFN and RBV results in a sustained virological response (SVR; defined as undetectable HCV RNA in the plasma 24 weeks after completion of therapy) in only 40%-50% of patients following 48 weeks of therapy.4-6 In patients with genotype 1 infection who failed to achieve SVR with a prior pegIFN/RBV regimen, retreatment with pegIFN and RBV for 48 weeks resulted in SVR rates ranging from 4% in nonresponders (did not achieve undetectable HCV RNA levels at any time during therapy) to 23% in relapsers (HCV RNA undetectable at end of treatment but returned following discontinuation of treatment).7 PegIFN and RBV therapy is also associated with substantial side effects, including fatigue, headache, myalgia, fever, nausea, insomnia, and anemia.4-6, 8 Such side effects often necessitate discontinuations or dose reductions, which decreases the probability of achieving an SVR. Consequently, there is an urgent clinical need for more effective and better-tolerated anti-HCV therapies that can achieve higher SVR rates with shorter treatment duration.9, 10
A number of direct-acting anti-HCV agents,9, 10 including the nonstructural protein 3/4A (NS3/4A) protease inhibitors and the nucleoside and non-nucleoside polymerase inhibitors, are being evaluated for use in combination with pegIFN and RBV. The addition of a direct-acting anti-HCV agent to pegIFN and RBV has been shown to significantly increase the proportion of patients achieving an SVR and may reduce the duration of therapy to 24 or 28 weeks in some HCV genotype 1-infected patients.11-13 Furthermore, direct-acting anti-HCV agents may ultimately be used in novel combination regimens that eliminate pegIFN and/or RBV. Such combination regimens may further improve SVR rates and address the safety and tolerability concerns associated with the current standard of care.
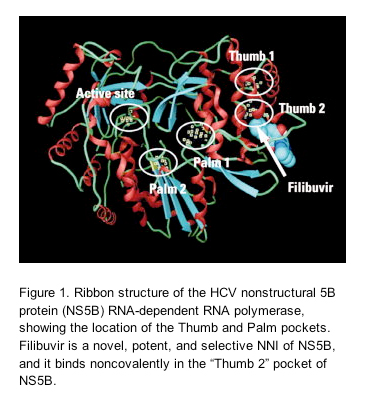
Filibuvir (formerly PF-00868554; Pfizer, Inc.) is a novel, potent, and selective non-nucleoside inhibitor (NNI) of the HCV nonstructural 5B protein (NS5B) RNA-dependent RNA polymerase, and it binds noncovalently in the "Thumb 2" pocket of NS5B (Fig. 1).14, 15In vitro, filibuvir is equipotent against genotype 1a and 1b replicons, with an overall mean median effective concentration (EC50) of 0.059 μM.16 In HCV-negative healthy volunteers, multiple oral doses of up to 300 mg three times daily (TID) administered over 14 days were safe and well tolerated and achieved plasma concentrations in excess of the in vitro protein binding-corrected EC50.15
Given the promising preclinical and clinical data, filibuvir was evaluated in HCV genotype 1-infected, treatment-naive (TN) and treatment-experienced (TE) patients. The primary objectives were to assess the antiviral activity, pharmacokinetics (PK), and safety and tolerability of multiple oral doses of filibuvir. This article presents data from two phase 1b studies: protocol number A8121002, NCT00445315 (study 1), and protocol number A8121006, NCT00671671 (study 2). This article also presents a brief summary of findings from the genotypic analysis of the NS5B nucleotide sequence from patients in studies 1 and 2.
RESULTS
Baseline Characteristics.
All 32 randomized patients in study 1 and all 20 patients in study 2 completed the studies. In study 1, there were no notable differences in demographic parameters or baseline HCV RNA concentrations between patients receiving filibuvir or placebo (Table 1). Demographics and baseline plasma HCV RNA concentrations were similar between studies 1 and 2. The genotypic distribution (1a versus 1b) within each cohort is listed in Table 1.
Antiviral Response.
Administration of filibuvir resulted in rapid, dose-dependent reductions in HCV RNA concentrations (Fig. 2). The mean maximum change from baseline in HCV RNA ranged from -0.97 log10 IU/mL (100 mg BID) to -2.30 log10 IU/mL (700 mg BID; Table 2). No patient achieved undetectable HCV RNA levels during filibuvir therapy. In study 1, the mean maximum reductions in HCV RNA concentrations were statistically greater compared with placebo (P < 0.05) for all doses of filibuvir evaluated. Because a placebo group was not included in study 2, an assessment of statistical significance could not be conducted. Dose-dependent increases in the proportion of patients in each dose group achieving a >2.0 log10 IU/mL maximum reduction in HCV RNA were observed, with none of the patients receiving 100 mg BID in study 1 and 9 of 10 (90%) patients receiving 450 mg BID in study 2 achieving a >2.0 log10 IU/mL maximum reduction in HCV RNA (Table 2).
Virologic breakthrough (>0.5 log10 IU/mL increase in HCV RNA from nadir) was observed before the end of treatment in 16 of 34 (47%) of patients who were treated for 8-10 days (Fig. 2; Table 2). In study 1, the frequency of virologic breakthrough was highest in the 300 mg BID group (67%), but was lower in the 300 mg TID and 450 mg BID groups (0% and 33%, respectively). To explore the hypothesis that the higher filibuvir exposures achieved by the 300 mg TID and 450 mg BID groups contributed to maintenance of viral suppression, the relationship between virologic breakthrough and filibuvir exposures (Cmin) was examined (Fig. 3). In each filibuvir dose group evaluated (all cohorts from study 1), patients in whom breakthrough occurred had filibuvir exposures similar to, or higher than, patients without breakthrough, indicating no relationship between filibuvir exposure and virologic breakthrough.
To determine the impact of HCV subtype on the antiviral activity of filibuvir, the data for all filibuvir doses that resulted in a >1.0 log10 IU/mL mean maximum reduction in HCV RNA were combined (all doses except placebo and 100 mg BID). The mean maximum change in HCV RNA for patients with genotype 1a or 1b was -2.06 log10 IU/mL and -2.14 log10 IU/mL, respectively (Table 2). In addition, the frequency of virologic breakthrough was comparable among patients infected with genotype 1a and 1b strains (Table 2).
Pharmacokinetics.
Absorption of filibuvir was rapid, with median Tmax ranging from 0.5-0.76 hours after dose for all groups in study 1 and cohort A of study 2, where filibuvir was administered under fasting conditions. The median Tmax was 2.0 hours in cohort B (study 2), where filibuvir was administered with food, indicating that food delays absorption. Following achievement of Cmax, filibuvir concentrations exhibited multiexponential decline with an apparent half-life ranging from 7.5-12 hours. In study 1, both median Cmax and AUC for filibuvir increased with increasing dose, with Cmax demonstrating more than proportional increases. Multiple-dose PK data suggested a small accumulation of filibuvir in plasma after BID and TID regimens. The mean accumulation ratio based on AUC for 100 mg BID, 300 mg BID, 450 mg BID, and 300 mg TID were 1.49, 1.15, 1.10, and 1.12, respectively. In study 2, the mean Cmax and AUC for filibuvir in cohort A were similar between days 1 and 10 (Table 3). In cohort B, the mean Cmax and AUC increased by ~29% and ~49%, respectively, between day 1 and 3 (Table 3).
Exposure-Response Modeling.
The Emax model appropriately described the relationship between filibuvir exposures and maximum reduction in HCV RNA concentration (Fig. 4) and confirmed the dose-dependency of HCV RNA reduction. The analysis suggests a plateau in the response to filibuvir and that increasing the filibuvir dose beyond 700 mg BID is unlikely to produce greater HCV RNA reductions. The log of baseline plasma HCV RNA concentration (normalized to 6) was identified as an influential covariate describing the Emax. There appeared to be no effect of genotype (1a versus 1b) on the Emax, E0, or AUC24,50 parameters (95% CI included null value). However, given that these studies were not powered to detect such differences, further exploration of the covariate-parameter relationships will be performed when new data emerge. The parameter estimates, their relative standard errors, and the associated 95% CIs are presented in Table 4.
Safety and Tolerability.
Filibuvir was well tolerated at all doses evaluated in these two studies. The most frequently reported AEs were headache, flatulence, and fatigue in study 1 (Table 5); headache and dyspepsia (four patients each) were reported in study 2, cohort A, and headache (three) and dry mouth (two) were reported in study 2, cohort B. There were no trends toward increasing frequency or severity of AEs with increasing doses of filibuvir. All AEs were mild or moderate in severity (one moderate AE in the 450 mg BID group in both studies). No temporary discontinuations or withdrawals due to AEs were required, and no serious AEs or deaths were reported. No clinically significant changes in vital signs, electrocardiogram parameters, or laboratory values were reported during treatment.
NS5B Sequence Analysis.
Mutations in NS5B at position Met423 were the preferred resistance pathway selected following filibuvir therapy. Before treatment, all patients were infected with virus encoding wild-type methionine at position 423 in NS5B. After treatment, virus from 29 of the 38 patients who received filibuvir >100 mg BID encoded amino acid variants at NS5B residue Met423. There was no significant difference in the frequency of appearance of position 423 mutations between subtype 1a (19 of 25; 76%) and subtype 1b (10 of 13; 77%) viruses (Fisher's exact test; P = 1.00). Mutations at residue 423 were consistently associated with virologic breakthrough (>0.5 log increase in HCV RNA from nadir) in patients receiving >100 mg BID. Sequence analysis of the day 28 follow-up samples indicated that reversion toward baseline methionine at position 423 was common (24 of 29 patients, 83%). One patient who received filibuvir 450 mg BID, who did not respond to treatment at all time points, was infected with a virus encoding an Arg422Lys variant.
Discussion
This is the first report of the antiviral activity and safety of filibuvir in HCV-infected patients. Data from these two phase 1b studies showed that filibuvir potently inhibited viral replication in a dose-dependent manner in patients infected with HCV genotype 1. The mean maximum reduction in HCV RNA concentrations ranged from -0.97 log10 IU/mL (100 mg BID) to -2.30 log10 IU/mL (700 mg BID). In study 1, the mean maximum reductions in HCV RNA were statistically greater than placebo for all filibuvir doses evaluated.
The 450 mg BID dose was investigated in TN patients (study 1) and TE patients (study 2) to assess any effect of prior treatment with pegIFN and RBV on the antiviral activity of filibuvir. When the nonresponder was excluded from the TN group, the maximum reduction in HCV RNA was not significantly different from that observed in the TE cohort in study 2, suggesting the antiviral activity of filibuvir is not affected by prior treatment status.
Previously published in vitro data demonstrate that the antiviral activity of filibuvir is comparable against the two most common subtypes of HCV genotype 1 (1a and 1b; mean EC50 versus 1a = 0.081 μM; mean EC50 versus 1b = 0.033 μM).16 In the present study, similar mean maximum reductions in HCV RNA were observed for 1a and 1b isolates (-2.06 and -2.14 log10 IU/mL, respectively). In addition, the frequency of virologic breakthrough was similar among patients infected with subtype 1a and 1b strains, and there was no significant difference in the frequency of appearance of position 423 mutations in patients infected with genotype 1a and 1b strains. The influence of genotype 1 subtype on maximal reduction in HCV RNA concentration was also tested in the exposure-response analysis, and it did not appear to have an effect. Therefore, these findings are consistent with in vitro data and further indicate that the antiviral activity of filibuvir is comparable against both subtype 1a and 1b strains of HCV.
Although administration of filibuvir resulted in significant decreases in HCV RNA concentrations during the first 72 hours of therapy, rebound was observed in some patients. In the 15 patients receiving >100 mg BID with virologic breakthrough (defined as >0.5 log increase in HCV RNA concentration), the breakthrough occurred after day 4. Longer treatment durations resulted in an increase in the frequency of virologic breakthrough with the 450 mg BID dose: two of six patients treated with 450 mg BID in study 1 (8 days of treatment) and 9 of 10 patients treated with 450 mg BID in study 2 (10 days of treatment).
In study 1, the frequency of virologic breakthrough was lowest in the 100 mg BID group, suggesting that the selective pressure exerted by this dose was insufficient to completely suppress replication of wild-type variants and enable the outgrowth of potentially less fit filibuvir-resistant variants. This observation is consistent with results from monotherapy trials with the HCV protease inhibitors boceprevir and telaprevir. In boceprevir monotherapy trials,19 patients who achieved a >2.0 log maximum reduction in HCV RNA were more likely to develop protease-inhibitor resistance mutations than those patients who achieved <2.0 log maximum reduction in HCV RNA. Similarly, in telaprevir monotherapy trials,20 higher telaprevir exposures were associated with an increased frequency of high-level resistant variants, indicating that the selection of resistant variants requires greater selective pressure.
Results from the exposure-response analysis suggest that increasing filibuvir doses beyond those tested in studies 1 and 2 is unlikely to result in greater reductions in HCV RNA concentrations. Based on the relationship observed for filibuvir dose and exposure (data not shown), doses in excess of 200 mg BID are expected to achieve 24-hour exposures resulting in at least half the maximal response, whereas doses in excess of 600 mg BID are expected to achieve exposures approaching the maximal response. A phase 2a study evaluating the effect of filibuvir given at 200, 300, and 500 mg BID (given for 4 weeks in combination with pegIFN and RBV) on HCV RNA concentrations showed that a greater proportion of patients achieved rapid virological response (>60%) at all filibuvir doses tested compared with the standard of care (0%).21 The exposure-response analysis, in conjunction with phase 2a combination study results, indicates that doses producing at least half the maximal response in monotherapy studies for filibuvir may be sufficient when used in combination with pegIFN and RBV to improve efficacy compared with current standard-of-care therapy.
Variants at NS5B residue 423 provided a clear correlate of virologic breakthrough in these clinical studies (P. Troke, M. Lewis, P. Simpson, K. Gore, J. Hammond, C. Craig, M. Westby, unpublished data, 2010).22 This finding is consistent with in vitro resistance data,16, 22 where high-level resistance has been demonstrated with variants (isoleucine, threonine, and valine) at residue 423. This finding is also consistent with data reported for other Thumb 2 NNIs. Specifically, variants at position 423 were identified via clonal sequence analysis as being the most predominant following VCH-759 exposure.23 There is no scientific rationale to expect that cross-resistance would occur between filibuvir and protease inhibitors and polymerase inhibitors that bind in other pockets in the polymerase protein.24
Several studies have been conducted to investigate the prevalence of known HCV drug-resistance mutations, including Met423Thr/Val/Ile, in the untreated HCV-infected patient population.25, 26 According to these studies, variants at position 423 are present in 2%-3% of the untreated patient population, and are associated with a reduction in the replicative fitness of the virus. The impact of pretreatment position 423 variants and/or the reduced fitness associated with these variants on response to therapy was not evaluated and is thus not yet understood. Although no position 423 variants were detected at baseline in either of the filibuvir monotherapy studies, a novel variant (Arg422Lys) was detected at baseline in virus isolated from a patient in study 1. This patient achieved a <0.5 log10 IU/mL reduction in HCV RNA concentration and was considered a nonresponder to filibuvir. According to the Los Alamos HCV database,27 this variant is uncommon in the HCV population, being present in just one of 352 genotype 1 NS5B sequences in the database.
The level of antiviral activity, resistance profile, and subtype 1a/1b activity observed for filibuvir in these studies compares favorably to other NNIs currently in development. Maximum reductions in HCV RNA reported for NNIs of HCV range from 0.6-3.7 log10 IU/mL,28 and the activity observed with filibuvir is well within this range. Many NNIs demonstrate differential antiviral activity against 1a and 1b subtypes. However, filibuvir, as well as other NNIs that target the Thumb 2 site of the enzyme (e.g., VCH-795),23 seem to demonstrate equivalent antiviral activity against 1a and 1b subtypes, which may be a function of the particular binding site. Safety or tolerability concerns associated with other NNIs under development, such as QT prolongation, gastrointestinal AEs, hepatotoxicity, and rash, were not observed in either of these filibuvir studies.
In conclusion, data from the two studies presented here show that filibuvir is a potent inhibitor of HCV replication in vivo and is well tolerated in HCV genotype 1-infected patients, supporting further clinical evaluation. Filibuvir is currently being evaluated in combination with pegIFN and RBV in treatment-naive patients.
Patients & Methods
All patients provided written informed consent before study participation. The protocol and informed-consent forms were approved by independent ethics committees at all study centers in accordance with national procedures. The studies were conducted according to the ethical principles in the Declaration of Helsinki and in compliance with all Good Clinical Practice guidelines, local laws, and regulations.17
In study 1, 32 TN patients were enrolled between January 2007 and May 2008 in three European centers: one in Belgium and two in Germany. In study 2, 20 patients were enrolled between April 2008 and December 2008 in a single center in Florida, USA. Both studies were conducted in chronic HCV genotype 1-infected patients, of either sex, aged 18-65 years with a body mass index of 18-34 kg/m2. Other key eligibility criteria included HCV RNA detectable in serum for ≥6 months and ≥100,000 IU/mL at screening. Women of childbearing potential or who were premenopausal were excluded, as were patients who were coinfected with hepatitis B or human immunodeficiency virus, who had evidence of severe or decompensated liver disease, or who had liver disease unrelated to HCV infection.
Study Design.
Study 1: This was a randomized, double-blind, placebo-controlled, sequential dose escalation study of orally administered filibuvir. Four cohorts of eight patients were randomized (6:2) to receive filibuvir (100, 300, or 450 mg every 12 hours [BID] or 300 mg every 8 hours [TID]) or placebo under fasted conditions for 8 days; treatments were given BID or TID on days 1 through 7, and once on day 8. The random allocation sequence used to assign patients was computer-generated. The sponsor generated the allocation sequence, and an investigator assigned participants to their groups sequentially as each patient was screened and in accordance with their randomization numbers. Patients and investigators were blinded but the sponsor was not.
Study 2: This was a nonrandomized, open-label, sequential group study of orally administered filibuvir in two cohorts. In cohort A, TE patients received filibuvir 450 mg every 12 hours (BID) for 10 days in a fasted state; in cohort B, TN patients received filibuvir 700 mg every 12 hours (BID) for 3 days in a fed state.
Antiviral Assessments.
Plasma HCV RNA concentrations were measured at screening, and in study 1 on day 0, days 1-3 (before dose, every 6 hours), daily over the treatment period, and at multiple time points during the follow-up period. In study 2, assessments took place on day 0, day 1 (predose and at 2, 6, 8, 12, and 14 hours after dose), daily over the treatment period, after the last day of dosing (day 11 for cohort A and day 4 for cohort B), and at multiple time points during the follow-up period. The HCV RNA was quantified using the Abbott RealTime HCV polymerase chain reaction assay according to manufacturer's instructions (lower detection limit of 12 IU/mL; Abbott Laboratories, Abbott Park, IL).
Pharmacokinetic Assessments.
In study 1, full PK profiles of filibuvir were obtained on days 1 and 8. Predose samples were collected on days 2 through 7. Starting on day 8, samples were collected up to 48 hours after dose. In study 2, full PK profiles were obtained on day 1 and following the last dose administered (day 10 for cohort A and day 3 for cohort B). Predose samples were obtained on days 2 through 9 for cohort A and days 2 and 3 for cohort B. Plasma concentrations of filibuvir were measured using a validated high-performance liquid chromatography-tandem mass spectrometric method (Bioanalytical Systems, Ltd., Warwickshire, UK).
Pharmacokinetic Noncompartmental Analysis Methods.
PK parameters were calculated by noncompartmental analysis of concentration-time data for days on which a full PK profile was obtained using internally validated PK analytical software (eNCA, Pfizer). The maximum observed concentration (Cmax) and the time to reach the Cmax (Tmax) were obtained directly from the data. AUC0-tau (area under the curve) over the dosing interval (0-tau, BID = 12 hours; TID = 8 hours) was estimated using the linear/log trapezoidal approximation.
Exposure-Response Analysis Methods.
Filibuvir exposures achieved over 24 hours (AUC24) derived from AUC0-tau obtained from noncompartmental analysis in individual patients in studies 1 and 2 were used to inform the exposure-response analysis of the maximum log change in HCV RNA concentration from baseline. Analysis was performed using a nonlinear mixed effects approach using the first-order conditional estimation (FOCE) method in NONMEM VI (Icon Development Solutions, Ellicott City, MD). The relationship was described by an Emax model as follows:
..............
where, MaxCFBi is the maximum observed log of HCV RNA drop relative to baseline (change from baseline) for individual i, AUC24i is the AUC over 24 hours for individual i after the final dose, E0 is the placebo response (at zero AUC24), Emax is the maximum attainable log of HCV RNA drop relative to baseline, and AUC24,50 is the AUC24 that gives half the maximal response.
The mixed effect model had an additive residual error component. The primary analysis of the effect of covariates on the model parameters was conducted by developing a full covariate model.18 The full model included the effect of baseline HCV RNA concentration on Emax and of genotype (1a versus 1b) on E0, Emax, and AUC24,50. This full model was then bootstrapped to obtain the 95% confidence intervals (CIs). The CIs were used to identify influential covariates based on the exclusion of either 0 (for continuous variable) or 1 (for categorical variable).
Safety and Tolerability Assessments.
All patients were evaluated for safety by physical examination, assessment of vital signs, electrocardiograms, and clinical laboratory tests at screening, baseline, and days 3, 7, and follow-up (day 28) in study 1; and screening, baseline, and days 7, 10, 15, and follow-up (day 28) for cohort A, and screening, baseline, and days 3, 5, and follow-up (day 28) for cohort B in study 2. Investigators recorded all observed or volunteered adverse events (AEs), as well as the severity (mild, moderate, severe) and the investigator's opinion of the relationship to study treatment. The AEs included adverse drug reactions, illnesses with onset during the study, and exacerbation of previous illnesses. In addition, the investigator recorded as AEs any clinically significant changes in physical examination findings and abnormal objective test findings (e.g., electrocardiogram results, laboratory test results, and so forth). The AEs were monitored throughout the study, and coded using MEdDRA version 11.0.
Statistical Analysis.
Baseline HCV RNA was defined as the average of screening and day 0 and 1 (0 hour) values. In study 1, data for patients receiving placebo across all cohorts and data for the two HCV subtypes were pooled for analysis. Change in log10 HCV RNA between baseline and day 8 and maximum change from baseline were secondary endpoints. In study 2, change in log10 HCV RNA between baseline and the last day of dosing, and maximum change from baseline, were coprimary endpoints. Statistical analyses were conducted at a two-sided 5% significance level. Descriptive statistics were used to summarize AEs.
HCV Genotype Determination.
HCV genotype was determined with the Versant HCV genotype assay, version 2.0 (LiPA; Bayer HealthCare Diagnostics, Tarrytown, NY) and confirmed retrospectively for all patients using phylogenetic analysis of NS5B nucleotide sequence at baseline (Lab21 Healthcare, Cambridge, UK). Genotype designations indicated in Table 1 are derived from retrospective phylogenetic analysis.
NS5B Sequence Analysis.
Two-step amplification and population sequencing of the HCV NS5B encoding region was performed on plasma samples collected at: screening and days 8 and 28 in study 1; at baseline and days 5, 10, and 28 for cohort A; and baseline and days 3, 5, and 28 for cohort B in study 2. The derived translations were aligned to reference sequences according to genotype: genotype 1a (Genbank accession, H77: NC_004102) and genotype 1b (Genbank accession, Con1: AJ238799).
|
|
| |
| |
|
|
|