| |
HIV Infection, Inflammation, Immunosenescence, and Aging - pdf attached
|
| |
| |
Download the PDF here
Annual Review of Medicine
Vol. 62: 141-155 (Volume publication date February 2011)
First published online as a Review in Advance on November 17, 2010
DOI: 10.1146/annurev-med-042909-093756
Steven G. Deeks
Department of Medicine, San Francisco General Hospital, University of California San Francisco
from Jules: studies show diet and exercise have an affect that can ameliorate the affects of accelerated or premature aging associated with HIV. Adequate & proper exercise and diet can reduce inflammation and have other affects that appear associated with accelerated aging; the reference is to vigorous aerobic exercise and resistance or weight lifting, and a mediterranean-type diet that reduces or minimizes animal fats and bad fats and optimizes intake of vegetables, fruits and low fat protein like fish.
"Many of the biologic factors that are thought to accelerate aging have also been implicated in the pathogenesis of HIV disease....... Visceral obesity is a well-established risk factor for many age-associated complications, including vascular disease and dementia. ........The fate of senescent cells may be a key determinant of health outcomes. Campisi and colleagues have argued that senescent cells often secrete inflammatory and other regulatory factors, resulting in chronic, low-level, "sterile" inflammation. These cells are known to accumulate in degenerating cells and may be causally associated with development of certain age-associated diseases......Chronic viral infections (e.g., HIV, CMV) cause excessive T cell turnover and the apparent accumulation of phenotypically senescent proinflammatory CD8+ T cells, as described......Mitochondrial toxicity......could theoretically contribute to cellular and tissue aging (89)......The double insult of aging and HIV to hematopoietic stem cells can contribute to many of the factors associated with immunosenescence, including reduced number of naive T cells, reduced T cell proliferation, and reduced ability of the immune system to mount an effective response to vaccines and infection........Chronic inflammation is strongly associated with the development of morbidity and mortality in the elderly and in those with HIV disease. Chronic viral infections such as the herpes and hepatitis viruses are an important cause of this persistent inflammation in both settings. CMV causes lifelong antigenic stimulation and the eventual development of an expanded population of well-differentiated, apoptosis-resistant, senescent T cells with limited proliferative potential (93, 94). The end result is an immune system with limited capacity to recognize novel antigens and hence prevent disease."
ABSTRACT
Although antiretroviral therapy for HIV infection prevents AIDS-related complications and prolongs life, it does not fully restore health. Long-term treated patients remain at higher than expected risk for a number of complications typically associated with aging, including cardiovascular disease, cancer, osteoporosis, and other end-organ diseases. The potential effect of HIV on health is perhaps most clearly exhibited by a number of immunologic abnormalities that persist despite effective suppression of HIV replication. These changes are consistent with some of the changes to the adaptive immune system that are seen in the very old ("immunosenescence") and that are likely related in part to persistent inflammation. HIV-associated inflammation and immunosenescence have been implicated as causally related to the premature onset of other end-organ diseases. Novel therapeutic strategies aimed at preventing or reversing these immunologic defects may be necessary if HIV-infected patients are to achieve normal life span.
Introduction
The development of antiretroviral therapy for the treatment of HIV infection is one of the greatest achievements of modern medicine. In a matter of a few years, the overall prognosis for HIV-infected patients shifted from years to decades. Although the initial regimens were complex and associated with significant short-term and long-term toxicity, current regimens are generally easy to administer, safe, and well tolerated. It is generally accepted that we are entering a phase of the epidemic in which we can achieve, and indefinitely maintain, control of HIV replication in the vast majority of patients.
Despite unquestioned success, combination antiretroviral therapy does not fully restore health. For reasons that remain poorly defined, long-term treated HIV-infected persons have an expected life span that is substantially shorter than that of their HIV-uninfected peers (1-4). This shortened life span is largely due to an increased risk of a number of "non-AIDS" complications, including heart disease, cancer, liver disease, kidney disease, bone disease, and neurocognitive decline. Many of these complications are similar to those observed among the elderly. Given the degenerative nature of most of these diseases, their impact on quality of life and function can be dramatic. These observations have led to growing concern that HIV-infected persons suffer from accelerated or premature "aging." This vaguely defined clinical scenario likely reflects a complex condition characterized by increased burden of comorbid diseases, higher prevalence of traditional behavioral risk factors (e.g., substance abuse), antiretroviral treatment toxicity, and chronic inflammation. These collectively result in functional decline and a higher than expected vulnerability to stressors or injury (5).
This review summarizes the complex epidemiologic, clinical, and pathogenesis data supporting the concepts that (a) antiretroviral-treated HIV-infected persons are at higher than normal risk for certain age-associated diseases, and (b) this risk is due in part to irreversible HIV-associated immunologic dysfunction. This review does not seek to summarize the related but distinct effect of aging on HIV infection and its management, a topic that has been well-reviewed elsewhere (5, 6).
RISK OF AGE-ASSOCIATED DISEASE IS HIGHER IN ANTIRETROVIRAL-TREATED HIV PATIENTS THAN IN HIV-UNINFECTED PERSONS
Several recent studies have attempted to determine the ability of modern antiretroviral treatment regimens to fully restore health. Each of these studies has significant limitations, including short-term follow-up, lack of proper adjustment for unmeasured confounders and the failure to consistently exclude patients who failed to achieve durable viral suppression. Despite these limitations, the data collectively strongly suggest that the risk of non-AIDS morbidity is higher among antiretroviral-treated HIV-infected individuals than their age matched uninfected peers for reasons directly related to the disease or its treatment (1-4).
Cardiovascular Disease
Most (but not all) studies have found higher rates of cardiovascular disease in HIV-infected populations than in age-matched HIV-uninfected populations (7). For example, in one large U.S. healthcare system, the risk of a myocardial infarction was much higher in HIV-infected versus uninfected persons (8). Similar results have been reported by other groups using either clinical events or well-validated surrogate markers as the outcome measure (e.g., carotid intima thickening, brachial artery flow-mediated dilation) (7-9).
The mechanism that accounts for the higher than expected rates of cardiovascular disease is the focus of intense investigation. People with HIV often have more traditional risk factors for heart disease (e.g., hypertension, diabetes, dyslipidemia), but these factors do not account for all of the increased risk (8, 10, 11). Abacavir and the various protease inhibitors have been associated with cardiovascular toxicity (12, 13). Because HIV-associated biomarkers such as CD4+ T cell nadir, proximal CD4+ T cell count, and markers of inflammation consistently predict an elevated risk of cardiovascular disease independent of other factors (14, 15), it is almost certain that HIV infection contributes to the elevated risk of cardiovascular disease. Our observation that the HIV-infected persons who are able to durably control HIV infection in the absence of therapy ("elite controllers") have more carotid disease than age-matched uninfected persons also argues for an effect of HIV-associated factors that is independent of direct toxicity, high viral replication, and advanced immunodeficiency (16).
Cancer
Another emerging dataset suggests that HIV infection is associated with a higher than expected rate of many non-AIDS cancers (17, 18). This risk is particularly evident for those non-AIDS-defining cancers that are known or believed to be caused by chronic infections (e.g., anal cancer, Hodgkin's disease, liver cancer), while the risk of other cancers (e.g., lung, colorectal, melanoma) is only slightly higher. This higher cancer rate is apparent even among long-term antiretroviral-treated patients, and it is strongly related to the degree of immunodeficiency-as defined by the on-therapy CD4+ cell count (19). Because the spectrum of cancers among HIV-infected persons is similar to that in the post-transplantation population (20), it has been argued that HIV-associated immune dysfunction may be the primary factor driving any excess risk.
Frailty
Aging is often defined on the basis of functional capacity rather than the collection of age-associated diseases (21). Aging is hence quantified by a series of metrics that cover specific domains. One such domain is frailty, which includes measures of sarcopenia (loss of muscle mass), osteoporosis, and muscle weakness. Although frailty has not been formally measured in HIV-infected populations, an estimate of frailty (the "frailty-related phenotype") was determined using interview data collected in the Multicenter AIDS Cohort Study (MACS). During the course of the study, about 14% of the HIV-infected population met the study definition of frailty at least once, whereas only 2% of the HIV-uninfected persons met this definition. The risk was most apparent after prolonged HIV infection and was strongly predicted by the peripheral CD4+ T cell count (in both treated and untreated individuals) (22). Since clinically apparent frailty as seen in the very old is likely uncommon in younger HIV-infected individuals, it has been proposed that other, more subtle measures of functional capacity be used in future studies (5).
Liver, Kidney, and Bone Disease
HIV-infected persons have a higher risk of both liver and kidney disease than age-matched uninfected persons (23, 24). Among HIV-infected persons, untreated disease (or persistent viral replication) is associated with a higher risk than treated disease, suggesting that HIV replication directly or indirectly harms these organs (25). The extent of viral replication appears to be a strong determinant of kidney disease (26, 27), while the peripheral CD4+ T cell count may be a more important determinant of either kidney or liver disease (14, 28). The extent to which effective antiretroviral therapy normalizes liver and kidney function is unknown and may be difficult to discern given that many antiretroviral drugs are directly toxic to these tissues.
In some studies, the prevalence of osteopenia and osteoporosis is at least three times higher in HIV-infected persons than HIV-uninfected controls (29). Fractures are also more common in HIV-infected persons (30). Persistent inflammation during therapy may be causally related to disease, as many of the inflammatory markers known to be higher in HIV disease have been associated with bone disease in HIV-uninfected persons (31). Other factors-including antiretroviral drug toxicity and vitamin D deficiency-also contribute to bone disease.
Neurologic Complications
The harmful effect of untreated HIV on peripheral and central nervous system (CNS) function was apparent very early in the epidemic. HIV-associated inflammation is believed to be a central factor in CNS disease. Effective therapy clearly prevents and often reverses this process, but residual disease often persists (32, 33). One of the most contentious issues in clinical HIV medicine is whether harm continues to accumulate during therapy and, if so, whether this ongoing harm is due to inadequate penetration of certain drugs into the CNS (thus allowing ongoing viral replication) or to residual inflammation (34, 35). Although persistent defects noted in treated patients are often subtle and of unclear clinical relevance, even subtle increases in the rate of progression could over time result in the early onset of clinically relevant conditions such as dementia.
AGING OF THE IMMUNE SYSTEM (IMMUNOSENESCENCE)
As with any organ system, the immune system exhibits characteristic changes as people get older. These changes are most apparent (or at least most studied) in T cells. Compared to younger adults, the immune system in older adults is marked by a number of characteristics, including (a) reduced number and function of hematopoietic stem cells, (b) thymic involution, (c) reduced circulating naive T cells, (d) increased frequencies of well-differentiated memory CD28- T cells with limited proliferative potential, (e) increased levels of many proinflammatory cytokines, including interleukin (IL)-6 and TNFα, and (f) decreased CD4/CD8 ratios (36, 37). Although most of these changes pertain to the adaptive immune system, other aspects of the immune system-including NK cells-exhibit reduced function in the very old, but data are limited. The few mucosa-based studies suggest that age-associated changes in peripheral blood are comparable to those seen in tissues.
These age-associated changes in immune function are often referred to as immunosenescence, a vaguely defined condition that refers to the age-associated changes in the immune system that are associated with morbidity and mortality. Among Swedish octogenarians and nonagenarians enrolled in a small, pathogenesis-oriented, population-based longitudinal cohort (the OCTO Immune Longitudinal Study), an inverted CD4/CD8 ratio was associated with short-term mortality (38). Comparable findings have been observed in other small cohorts (39). Other parameters from the OCTO cohort (and the subsequent NONA cohort) that predicted morbidity and mortality included reduced T cell proliferation, increased frequency of CD28- T cells and increased IL-6 (38, 40). The rare individuals who are able to survive to 100 or more years of age often lack these immunologic abnormalities (41).
The optimal T cell response is characterized by dramatic clonal expansion and generation of effector responses. The initiation of the T cell response requires interaction of antigen with the T cell receptor and at least one potent costimulatory receptor. Perhaps the most important costimulatory molecule is CD28, which is gradually downregulated as central memory cells differentiate into effector cells. The resulting CD28- cell population has shorter telomeres and is less able to proliferate. Many of these cells rapidly die, but some may become apoptosis resistant and long-lived (although in vivo data regarding the turnover of these cells are limited). These so-called senescent cells are proinflammatory and hence may have a viable effector function, but their expansion can contribute to heightened systemic inflammation and collateral harm (42). Because chronic antigen exposure and inflammation result in the gradual expansion of these CD28- cells, it is not surprising that chronic viral infections are associated with a progressive expansion of these cells.
Cytomegalovirus (CMV) infection may prove to be very instructive with regard to the mechanism whereby HIV causes the premature onset of age-associated diseases. Among the very old, CMV seropositivity is associated with dramatic expansion of CD8+CD28- T cells, with many of these cells directed at CMV (43). This remodeling of the immune system is associated with vaccine unresponsiveness, cardiovascular disease, and mortality (44-46).
HIV INFECTION AND IMMUNOSENESCENCE
Many of the T cell abnormalities associated with aging are similar to those observed in untreated HIV infection (42, 47, 48). These similarities are based on a diverse set of isolated nondefinitive observations, and hence the superficial similarities summarized here should be used to justify more intense investigation. Untreated HIV-infected adults and the elderly often exhibit low CD4/CD8 ratios, low naive/memory ratios, reduced T cell repertoire, reduced responsiveness to vaccines, and an expansion of CD28- effector T cells (see Table 1) (49, 50). The degree to which long-term antiretroviral therapy reverses these HIV-associated changes in the T cell compartment is the focus of ongoing investigation. In one recent study of treated HIV-infected adults and both young and old uninfected controls, the T cell phenotype (CD57+CD28-CD8+ T cells, naive/memory T cell ratios, activated T cells) of the HIV-infected cohort was more similar to the much older uninfected cohort (48).
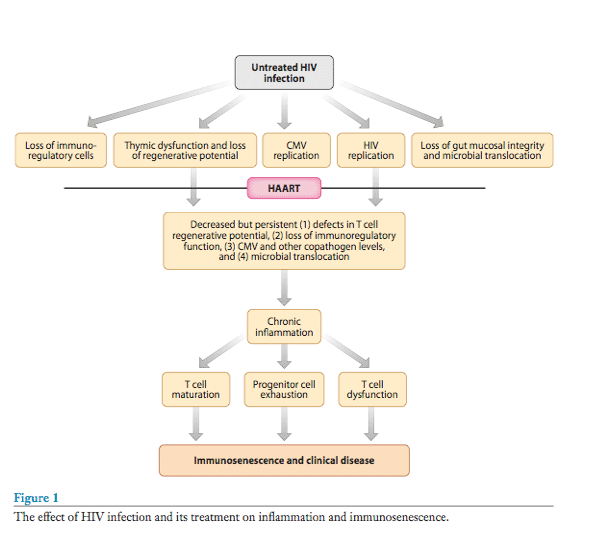
Many of the T cell characteristics associated with immunosenescence-including thymic dysfunction, T cell activation, and a reduced T cell regenerative potential-are more common among individuals who fail to exhibit robust CD4+ T cell gains during therapy than among those who achieve a normal CD4+ T cell count (51-53). Because a low CD4+ T cell count on therapy is a consistent proximal predictor of non-AIDS morbidity (54), these observations collectively suggest that HIV-associated immunosenescence contributes to persistent immunodeficiency and the early onset of age-associated diseases (see Figure 1). Focused investigation regarding this hypothetical pathway could lead to novel therapeutic interventions for both HIV-infected persons and the elderly.
The clinical significance of immunosenescence is often explored using vaccine responsiveness as the outcome. Effective antiretroviral therapy improves vaccine responsiveness, but residual defects remain, particularly if therapy is initiated late in the disease course. For example, among a cohort of 29 long-term treated patients (all of whom had high CD4 T cell counts), lymphoproliferatve responses to KLH, tetanus, and diphtheria toxoid were lower than those observed in HIV-seronegative controls, and predicted by both CD4 nadir and percentage of CD28-CD4+ T cells (but not current CD4) (55).
Untreated HIV infection is associated with persistently high levels of inflammation, as defined by levels of inflammatory cytokines such as IL-1β, IL-6, and TNFα. The coagulation system is also activated (23). Most if not all of these markers of inflammation decline with combination antiretroviral therapy, indicating that active HIV replication is either directly or indirectly responsible for this inflammatory response. Notably, the level of inflammation-as defined by IL-6, C-reactive protein (CRP), cystatin C and D-dimers-remains elevated despite durable and possibly complete suppression of HIV replication with antiretroviral therapy. This persistent inflammation during therapy is probably due to a number of factors, including ongoing HIV production (if not HIV replication), increased copathogen load (particularly CMV, but likely other herpesviruses as well), translocation of lipopolysaccharide (LPS) across a damaged gut mucosa, loss of T regulatory cells and other immunoregulatory cells, and the irreversible fibrosis of the thymus and lymphoid infrastructure (see Figure 1) (56-59).
The association between HIV infection and inflammation shares many similarities with the association between advanced age and inflammation. Indeed, many of the markers now being studied in HIV disease were first validated in cohorts of older individuals (60, 61), and many of the mechanisms thought to be causally associated with inflammation in HIV disease are also thought to be causally associated with the inflammation of aging (56). The strength of the association between certain inflammatory markers (e.g., IL-6, D-dimers) and risk of age-associated diseases and mortality is very strong in both patient groups (62), and generally higher than that seen in younger HIV-uninfected populations.
BIOLOGY OF AGING
From a biologic perspective, aging is typically defined as the progressive deterioration in physiologic function that occurs as a consequence of cumulative molecular, cellular, and organ damage. These changes invariably result in an increased susceptibility to disease, decreased responses to stress, and death. The clinical manifestations of this process include loss in end-organ function (e.g., liver, kidney, heart), bone loss (osteoporosis), muscle wasting (sarcopenia), neurocognitive decline, and loss in immunologic function (immunosenescence). There is a subtle distinction, however, between aging and age-associated diseases. The former is not generally considered a disease but rather a normal and well-conserved evolutionary process that is highly regulated and affects all organ systems. The latter is a series of clinical syndromes that increase in frequency as we age and are ultimately what cause death (to a purist, one cannot die of "old age"). This distinction can be confusing as many of the mechanisms known to regulate the aging process have been implicated in the pathogenesis of specific age-associated diseases. Also, the accumulation of comorbid diseases may be causally associated with accelerated aging, defined biologically or clinically.
Genetics of Aging
The rate at which a person ages is defined in part by his or her genetic background. Studies performed on elderly Danish twins in the modern era suggest that about 25% of the variability in life expectancy can be attributed to genetic factors (63). The genetic basis for these observations has been the focus of extensive research. To date, only the defective lipid carrier apolipoprotein E4 (apoE4) gene and the FOXO transcription factors (see below) have been consistently associated with longevity (64). Mutations that influence the expression of insulin-like growth factor (IGF) receptor are enriched in Ashkenazi Jewish centenarians (65). Because many of the genes known to increase longevity in animal models may result in a higher risk of cancer (by preventing cell death) and/or reduced fertility (by shifting resources from reproduction and growth to maintenance), it is likely that strong evolutionary pressures are acting to prevent exceptional longevity (66).
Notably, the harmful apoE4 gene has been associated with mortality in HIV disease (67), whereas the protective transcription factor FOXO3a has been associated with central memory T cell persistence in HIV infection. FOXO3a may represent one mechanism whereby certain individuals are able to remain healthy without antiretroviral therapy for years (68).
Molecular Biology of Aging
The best-characterized external (and hence potentially modifiable) factor associated with healthy aging is moderate caloric restriction. In nearly all species studied to date, experimental restriction of caloric intake to levels below that when fed ad libitum but above that which causes starvation is associated with increased longevity (69, 70). In a recently published study that took decades to complete, rhesus monkeys randomized to a 30% reduction in caloric intake exhibited a reduced risk of dying from age-associated diseases (although this mortality benefit was not significant when a large number of non-age-associated deaths were included in the analysis) (71). Caloric restriction in these monkeys reduced the risk of cancer, diabetes, and heart disease. Caloric restriction may also enhance T cell function and prevent immunosenescence in aging nonhuman primates (72). Whether this approach will work in humans is not known because such diets are nearly impossible to maintain; however, in a recent short-term prospective clinical trial, calorie restriction resulted in reduced energy expenditure, increased mitochondrial content, and increased expression of many genes associated with mitochondrial function and longevity (73).
Many of the experimental mutations that prolong life expectancy affect the caloric and nutrient signaling pathways, providing definitive proof for the role of diet in aging. Genetic mutations that decrease the activity of the insulin/IGF pathway equivalents in C. elegans double the life spans of these worms. The FOXO transcription factors regulate a wide range of genes known to be involved in the stress response and are critically important in mediating the antiaging effects of caloric restriction and reduced insulin/IGF in worms (74). Similar albeit less dramatic observations have been made in flies and mice. The apparent enrichment of certain genetic mutations within the insulin and IGF pathways among human centenarians strongly suggests that the ability of these crucial pathways to regulate longevity is conserved from worms to humans (74).
The target of rapamycin (TOR) is also involved in regulating the cellular response to nutrients and is a key component of the insulin/IGF pathways. Activation of TOR in many experimental models results in a shift in metabolism toward growth and reproduction, whereas inhibition of this enzyme results in a number of outcomes associated with cell maintenance, including higher levels of autophagy (i.e., the recycling of digested cellular components) (75, 76). TOR is of high interest clinically because the administration of low doses of rapamycin-an immunosuppressant approved by the U.S. Food and Drug Administration to prevent transplant rejections-prolongs the life of various species, including mice (77).
The silent information regulator protein deacetylases (sirtuins) are yet another family of proteins that influence the aging process. The sirtuins regulate many aspects of cellular metabolism, and therapeutic activation of at least one sirtuin (SIRT1) in experimental models is associated with reduced activity of cellular activation (via NFκB and other regulatory enzymes) and prolonged life span. The impact of caloric restriction on health may be mediated via the sirtuin family (73).
Cellular Biology of Aging
Normal cells cannot proliferate indefinitely. After multiple rounds of cell division, an irreversible state of replicative senescence occurs (the "Hayflick Limit"). This phenomenon is regulated in part by the progressive shortening of telomeres, the repeated DNA sequences (TTAGGG) that cap the ends of the chromosomes (78). In addition to stimulating specific pathways that influence the cell cycle (79), telomere length also influences the activity of p53 tumor suppressor pathways, resulting in either apoptosis or cell senescence and the prevention of malignant transformation. Genomic damage and mitochondrial damage-which are caused by many environmental exposures that are common in HIV infection-also activate many of these pathways, leading to either apo-ptosis or cell senescence and the prevention of cancer (80, 81).
The fate of senescent cells may be a key determinant of health outcomes. Campisi and colleagues have argued that senescent cells often secrete inflammatory and other regulatory factors, resulting in chronic, low-level, "sterile" inflammation. These cells are known to accumulate in degenerating cells and may be causally associated with development of certain age-associated diseases (81). Although these observations have largely focused on stromal and epithelial cells, they may also apply to T cells and other immune cells. Chronic viral infections (e.g., HIV, CMV) cause excessive T cell turnover and the apparent accumulation of phenotypically senescent proinflammatory CD8+ T cells, as described above.
BIOLOGY OF AGING AND ITS IMPLICATIONS FOR THE PATHOGENESIS OF NON-AIDS MORBIDITY DURING ANTIRETROVIRAL TREATMENT
Many of the biologic factors that are thought to accelerate aging have also been implicated in the pathogenesis of HIV disease. It is the central hope of this review that these two distinct fields of study could merge, as knowledge gained in one could accelerate progress in the other.
Visceral Fat, Insulin Resistance, and the Metabolic Syndrome
HIV infection and/or its treatment may cause peripheral fat wasting (lipoatrophy) and central fat gain. Visceral obesity is a well-established risk factor for many age-associated complications, including vascular disease and dementia. Visceral obesity is also a well-known source for many of the chronic inflammatory proteins known to influence both aging and HIV disease outcomes (82). Finally, visceral obesity is a strong predictor of insulin resistance, which is common in HIV-infected patients and is a strong determinant of aging (83, 84). Development of therapeutic agents aimed at reversing the complex effect of HIV infection on visceral fat, insulin resistance, and the lipodystrophy syndrome is the focus of intense investigation.
Genotoxicity and Mitochondrial Dysfunction
DNA damage and telomere shortening are strong determinants of cellular aging; each can activate the p53 and other tumor suppressor pathways, leading to apoptosis or cellular senescence and the inability to maintain tissue homeostasis (81, 85). Mitochondria dysfunction may also contribute to cellular aging, either by the release of potentially harmful reactive oxygen species (86) or by directly activating p53 and similar pathways. Release of mitochondrial products into the circulation may result in harmful levels of inflammation (87).
Zidovudine, stavudine, and perhaps other nucleoside analogs inhibit mitochondria synthesis, cause the release of mitochondrial DNA, and increase the risk of oxidative damage. Mitochondrial toxicity is thought to be a major contributor to fat redistribution and other metabolic abnormalities that are commonly seen with certain antiretroviral drug regimens (88). It has also been suggested that certain nucleoside analogs might inhibit telomerase (which is a reverse transcriptase); this could theoretically contribute to cellular and tissue aging (89).
T Cell Regenerative Failure
A reduced ability to regenerate T cells is another feature common to both HIV disease and advanced age. The progressive loss of hematopoietic progenitor cells is likely a major factor in the normal aging process (90). Age-associated factors that may accelerate the loss of these cells include excessive turnover, damage to the microenvironment, exposure to oxidative stress, and the accumulation of genetic and epigenetic alteration. Cellular senescence or apoptosis is often the final outcome (91). HIV may be able to directly infect hematopoietic stems or may negatively affect their function via exhaustion and/or local damage to the stem cell microenvironment (92). The double insult of aging and HIV to hematopoietic stem cells can contribute to many of the factors associated with immunosenescence, including reduced number of naive T cells, reduced T cell proliferation, and reduced ability of the immune system to mount an effective response to vaccines and infection. A loss of these stem cells may also contribute to a reduction in the number of endothelial progenitor cells, which can contribute to vascular dysfunction and cardiovascular disease.
This same story could be told with regard to thymic function. Advanced age and HIV infection are both associated with a progressive and possibly irreversible loss of thymic function; this process in theory could contribute to progressive immunologic dysfunction (including chronic inflammation) and the development of many age-associated diseases (37, 92).
Inflammation
Chronic inflammation is strongly associated with the development of morbidity and mortality in the elderly and in those with HIV disease. Chronic viral infections such as the herpes and hepatitis viruses are an important cause of this persistent inflammation in both settings. CMV causes lifelong antigenic stimulation and the eventual development of an expanded population of well-differentiated, apoptosis-resistant, senescent T cells with limited proliferative potential (93, 94). The end result is an immune system with limited capacity to recognize novel antigens and hence prevent disease. Because copathogens are more common in people with HIV, and because they appear to have a deleterious immunologic and clinical impact in HIV disease (58, 95), it seems reasonable to postulate coinfections may contribute to the "accelerated aging" syndrome now being observed in HIV-infected individuals (96).
Conclusion
The consistent observation that HIV-infected persons have a higher than expected risk for a number of conditions commonly associated with aging has led to the widespread assumption that HIV accelerates the aging process. It should be emphasized, however, that it is impossible to truly define the independent effect of HIV infection on this risk, as infected and uninfected persons differ with regard to many important and difficult to measure factors that influence the risk of developing an age-associated complication. Certain biomarkers that predict morbidity in the very old are higher than expected in younger HIV-infected persons, and this consistent observation provides indirect evidence that HIV infection might accelerate the aging process.
A careful integration of the basic biology of aging with the biology of HIV infection may lead to novel insights into the larger questions of why people age and why antiretroviral therapy fails to restore health. An integration of these two disciplines could provide the rationale for the codevelopment of novel therapeutics. For example, the National Institute of Aging has sponsored a collaborative program aimed at identifying possible therapeutic agents that delay physiologic aging (97). Any drug that advances into clinical trials among the elderly might also be considered in younger antiretroviral-treated HIV-infected patients. Interventions that have shown promise as antiaging therapeutics in experimental systems include resveratrol, rapamycin, acetyl-L-carnitine and alpha-lipoic acid, telomerase activators, caloric restriction, and stem cell therapy (97). Approved drugs that have an anti-inflammatory effect and are often used in older adults-including aspirin, omega-3 fatty acids, vitamin D, and the statins-are now being studied as adjuncts to antiretroviral therapy in younger HIV-infected individuals. It is expected, but not yet the focus of prospective investigation, that lifestyle modification-including moderate exercise and dietary changes-might also prove very beneficial as an adjunct to standard antiretroviral regimens.
| |
| |
| |
|
|
|