| |
Intrinsic HIV/antiviral immunity: recent advances in understanding of the roles of intrinsic antiviral factors that restrict infection by HIV and influenza virus
|
| |
| |
A quantitative basis for antiretroviral therapy for HIV-1 infection by R Siliciano et al - (02/23/12)
"SAMHD1 is antiviral...."
"understanding intrinsic antiviral factors-immunity/innate immune sensors-mechanisms of action.....This line of research may be key to the development of effective treatments for HIV and other devastating pathogens."
"The repeated failure to produce an effective HIV vaccine is a humbling reminder that full understanding of the immune response to HIV is still lacking. In fact, very little is known about innate immune responses to retroviruses in general"
"The rapid progress in innate immunity research in the past decade has been breathtaking. Several families of innate immune sensors have been discovered and the signaling pathways that they trigger are being rigorously investigated. By comparison, less is understood of the intrinsic antiviral factors and their mechanisms of action. The intense medical interest in HIV, influenza virus and other viruses has begun to elucidate some of the host restriction factors that limit infection by these viruses. Such studies have also provided striking examples of the 'arms race' between the host and virus during their coevolution. Further studies of individual viral proteins will continue to shed light on the host immunity that attacks various viruses. Conversely, investigation of host antiviral factors will elucidate how viruses evade the host's immunosurveillance. Studies of viral tropism in certain cell types have had an excellent track record of discovering important antiviral factors, such as APOBEC3G, TRIM5a, tetherin and SAMHD1. There is no reason to think that such a successful track record will end any time soon."
-----------------------------
Nature Immunology | Review March 2012 issue
Intrinsic antiviral immunity
Abstract
Intrinsic antiviral immunity refers to a form of innate immunity that directly restricts viral replication and assembly, thereby rendering a cell nonpermissive to a specific class or species of viruses. Intrinsic immunity is conferred by restriction factors that are mostly preexistent in certain cell types, although these factors can be further induced by viral infection. Intrinsic virus-restriction factors recognize specific viral components, but unlike other pattern-recognition receptors that inhibit viral infection indirectly by inducing interferons and other antiviral molecules, intrinsic antiviral factors block viral replication immediately and directly. This review focuses on recent advances in understanding of the roles of intrinsic antiviral factors that restrict infection by human immunodeficiency virus and influenza virus.
Introduction
Primordial forms of antiviral immunity: RNAi and CRISPR
One of the earliest forms of antiviral immunity in eukaryotic evolution is RNA-mediated interference (RNAi). RNAi is the predominant mechanism of antiviral defense in plants and invertebrates, and it is also a primordial form of immunity to viral infection in vertebrates. Infection by RNA viruses leads to the generation of long double-stranded RNA (dsRNA) that is structurally different from host cellular RNA, which is single stranded with short and often imperfectly matched stem loops1, 2. In plants and invertebrates, the endoRNase Dicer cleaves long viral dsRNA and gives rise to small interfering RNA (siRNA) duplexes. These siRNAs are then loaded into the RNA-induced silencing complex to target viral mRNA or genomic RNA for degradation, thereby inhibiting viral replication (Fig. 1a). In plants and nematodes, but not in insects, the antiviral RNAi response is amplified by RNA-dependent RNA polymerases that replicate the incoming viral RNA, which can then be processed by Dicer to generate more siRNA3, 4, 5. Inhibition of RNAi in plants increases their susceptibility to many plant viruses6, 7. To counteract antiviral RNAi, many plant and invertebrate viruses have evolved suppressor-of-RNA-silencing proteins that are important for establishing infection8, 9.
An even more ancient form of antiviral immunity is the CRISPR system (clustered, regularly interspaced short palindromic repeats) that protects bacteria and archaea from bacteriophages and conjugative plasmids10. In this system, some of the invading DNA sequences from bacteriophages or plasmids are acquired and integrated into the CRISPR loci of the host as repeat elements. The DNA repeats in the CRISPR loci are transcribed and processed into siRNA (crRNA) by the bacterial CRISPR-associated proteins. The crRNAs are incorporated into large CRISPR-associated protein complexes (such as Cascade or Cas6), which then degrade the invading viral DNA in a sequence-specific manner that is guided by the crRNA. This bacterial antiviral mechanism resembles RNAi in that siRNAs are used to guide the destruction of invading nucleic acids with a high degree of sequence specificity. However, there are also clear differences; for example, the precursors of crRNA are single-stranded RNA and the targets of destruction by crRNA are viral DNA. The CRISPR system has not been found in eukaryotic cells.
Extensive effort has been made to try to demonstrate antiviral RNAi responses in vertebrates, especially in mammalian cells11. Most of these efforts have failed to recover siRNAs of viral origin in mammalian cells infected with a variety of RNA and DNA viruses12. DNA viruses such as herpesvirus do produce small RNA such as microRNA, but not siRNA, and viral microRNA has an important role in establishing infection13. Vertebrates have a more versatile interferon system than the RNA-based immunity of plants and invertebrates; this interferon system constitutes an elaborate protein-based antiviral immunity (Fig. 1b). This evolutionary 'upgrade' is important for vertebrates to cope with more complex pathogens and the diversity of nucleic acids introduced into the cell and to minimize off-target effects of RNAi on host mRNA. However, vertebrates do retain evolutionary 'fossils' of the antiviral RNAi machinery. For example, mouse embryonic stem cells express endogenous siRNA similar to the antiviral siRNA found in plants and invertebrates, although it is unclear whether this siRNA targets any genes or has any important defensive role. Long dsRNA can induce sequence-specific RNAi against target mRNA in mouse embryonic stem cells. These mammalian embryonic stem cells lack functional interferon signaling pathways, which might explain why they have retained remnants of the antiviral RNAi machinery14, 15.
In addition to using antiviral RNAi, invertebrates such as Drosophila melanogaster have evolved the Toll signaling pathway that is important for both antimicrobial defense and development of the embryo16. The Drosophila protein Toll is the founding ortholog of the mammalian Toll-like receptors (TLRs) that are critical for innate immune responses to pathogens. In Drosophila, infection with Gram-positive bacteria or fungi activates the Toll pathway, which leads to the production of antimicrobial peptides, whereas mammalian TLRs trigger proinflammatory and interferon responses16. The remarkable similarity between Drosophila Toll and mammalian TLR signaling pathways and the greater complexity of the latter underscore the evolutionary requirements for more intricate antiviral immunity in mammals. Drosophila also activate the so-called Imd (immune-deficiency) pathway to induce antimicrobial peptides in response to infection with Gram-negative bacteria. This pathway resembles the tumor-necrosis factor pathway in mammals in that both use similar mechanisms to activate signaling molecules of the IKK and MAPK families.
The vertebrate interferon response
Vertebrates are constantly challenged by potentially pathogenic microbes that can introduce a variety of proteins and nucleic acids into the cell. To counter this, vertebrate cells express many different pattern-recognition receptors (PRRs) that can detect the pathogen-associated molecular patterns of viruses and other microbes, which in turn activate antiviral interferon and proinflammatory responses17, 18. Through the secretion of interferon, the response can be amplified and spread to surrounding uninfected cells via the Jak-STAT signaling pathway and thereby activate hundreds of interferon-stimulated genes (ISGs), most of which encode products with profound antiviral effects, such as the degradation of viral nucleic acids or inhibition of viral gene expression19 (Fig. 1b).
PRRs are proteins that recognize the molecular patterns of microorganisms and trigger innate immune responses to limit microbial infection20, 21. Mammalian PRRs include TLRs, RIG-I-like receptors (RLRs), Nod-like receptors (NLRs) and C-type lectin receptors. These receptors activate signaling cascades that lead to activation of the transcription factors NF-kB and AP-1, which induce proinflammatory cytokines. During viral infection, viral nucleic acids are the main pathogen-associated molecular patterns detected by the host innate immune receptors, which include RLRs in the cytosol and a subfamily of TLRs that localize to the endosomal membrane. TLR7 and TLR9 detect viral RNA and DNA, respectively, in the endosomal lumen of virus-infected cells. These TLRs contain a TIR domain that recruits the adaptor MyD88 from the cytoplasm. MyD88 in turn recruits the kinases IRAK1 and IRAK4 and the ubiquitin E3 ligase TRAF6 (ref. 22). TRAF6 activates the kinase complex IKK, which phosphorylates the NF-kB inhibitor IkB. This phosphorylation targets IkB for degradation by the ubiquitin-proteasome pathway, thereby allowing NF-kB to enter the nucleus to turn on inflammatory genes. MyD88 and TRAF6 on the endosomal membrane also recruit another transcription factor, IRF7, which is phosphorylated by IKKa and then enters the nucleus to induce type I interferons, especially interferon-a (IFN-a; Fig. 1b).
RLRs include RIG-I, Mda5 and LGP2, all of which share a RNA helicase domain that contains a DExD/H (Asp-Glu-X-Asp/His, where 'X' is any amino acid and 'Asp/His' indicates either aspartic acid or histidine) box23. RIG-I and Mda5 contain two caspase-recruitment domains (CARDs) in tandem at their amino terminus that are important for their signaling functions. LGP2 lacks the CARDs needed for signaling and probably has a regulatory role. RIG-I also contains a carboxy-terminal domain that binds to viral RNA containing 5'-triphosphates24, 25. The binding of viral RNA to the carboxy-terminal domain of RIG-I induces a conformational change that exposes the amino-terminal CARDs, which recruit the ubiquitin E3 ligase TRIM25 to catalyze the synthesis of Lys63 (K63) polyubiquitin chains26, 27, 28. These ubiquitin chains bind to and activate RIG-I CARDs26, which then interact with the CARD of the signaling adaptor MAVS (also known as IPS-1, VISA or CARDIF)29, 30, 31, 32. This interaction promotes the aggregation of MAVS into microfibrils through a prion-like mechanism33. MAVS aggregates on the mitochondrial membrane, then recruits signaling proteins from the cytoplasm, which leads to the activation of IKK and the IKK-like kinase TBK1. TBK1 phosphorylates the transcription factor IRF3, which causes IRF3 to dimerize and translocate to the nucleus, where it functions together with NF-kB to induce IFN-b and other antiviral molecules (Fig. 1b). Genetic experiments have demonstrated that RIG-I is essential for immune defense against many RNA viruses, including paramyxoviruses (such as Sendai virus and Newcastle disease virus) and orthomyxoviruses (such as influenza virus) and some positive-stranded RNA viruses (such as hepatitis C virus and Japanese encephalitis virus). In contrast, Mda5 is required for interferon induction by picornaviruses (such as encephalomyocarditis virus)34. The viral ligands for Mda5 have not been precisely defined but are thought to consist of long dsRNA containing branched structures35. MAVS is required for interferon induction by both RIG-I and Mda5 (ref. 36).
DNA viruses can also induce interferons through the endoplasmic reticulum membrane adaptor STING (also known as MITA, MPYS or ERIS)37. Many proteins have been proposed to detect double-stranded DNA in the cytosol; these include DAI, IFI16 and DDX41 (refs. 38,39,40). In addition, RNA polymerase III detects AT-rich DNA in the cytosol and converts the DNA into 5'-triphosphate RNA, which can then activate the RIG-I pathway to induce interferons41, 42. It remains to be determined whether one of these proteins functions as a dominant sensor of cytosolic DNA in vivo or whether multiple sensors of cytosolic DNA exist and each functions in a distinct cell type to induce interferons.
In addition to inducing interferons, both DNA and RNA viruses can trigger cell death and induce inflammatory cytokines such as interleukin 1b (IL-1b) through activation of the inflammasome, which belongs to the NLR family43. Whereas viral RNA seems to activate the NLRP3 inflammasome to generate mature IL-1b, viral DNA is instead detected by the AIM2 inflammasome44, 45.
Intrinsic versus innate immune factors
Many host proteins in addition to those discussed above can also detect viral infection and exert antiviral activities. For example, the deaminase APOBEC3G edits the human immunodeficiency virus (HIV) genome to inhibit HIV replication, and the E3 ubiquitin ligase TRIM5a targets the incoming HIV capsid protein and modulates uncoating of the capsid46. These proteins, called 'intrinsic antiviral factors' here, can also be classified as PRRs because they directly bind to viral components. However, unlike TLRs and RLRs, which inhibit viral infection indirectly by activating signaling cascades that result in the transcription of genes encoding new antiviral factors such as interferons, intrinsic restriction factors inhibit viral replication directly, often before the onset of the interferon response. Thus, intrinsic antiviral factors preexist in certain cell types, although most of these factors can be further induced by interferons to amplify their antiviral activity. In this review, we will use the term 'intrinsic antiviral factor' exclusively for host factors that can directly recognize viral components and are able to block viral replication immediately. In contrast, 'innate immune factor' covers a much broader spectrum and refers to host factors such as TLRs, RLRs and NLRs, which participate in the recognition, signaling and orchestration of both innate and adaptive immune responses to viral infection.
Mechanistic studies of intrinsic antiviral factors are important because viruses almost always must express proteins or devise strategies that counteract these factors to replicate in the host cell. Moreover, the expression patterns of intrinsic antiviral factors often determine the permissiveness of a cell type to a virus (or to a mutant virus that lacks the counteracting viral protein). In many cases, this permissiveness versus nonpermissiveness holds the key to the discovery of intrinsic antiviral factors and parallel evasion mechanisms of the virus (discussed below). During evolution, many intrinsic antiviral factors are under strong positive selection through coevolution with the virus. In some cases, they have an important role in limiting cross-species transmission of a virus and thereby determining the viral tropism (discussed below).
Below we will discuss recently discovered intrinsic antiviral factors against HIV-1 and influenza virus. Two others, RNase L and PKR, are also important intrinsic antiviral factors that have been well characterized and extensively reviewed. The pathway of OAS (2'-5'-oligoadenylate synthetase) and RNase L was one of the first interferon-induced antiviral pathways discovered47. PKR mediates a multifaceted antiviral response by regulating protein translation by the ribosome and by promoting innate immune signaling48. These two factors target a broad spectrum of viruses and will not be discussed further in this review.
Intrinsic immunity to HIV
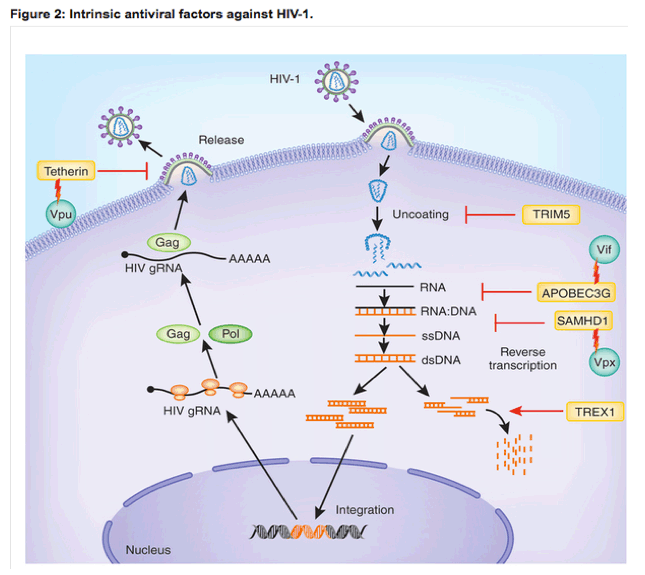
HIV enters T cells and macrophages via binding of the viral envelope protein gp120 to the coreceptor CD4, which enables the membrane-proximal portion of the envelope subunit gp41 to bind to the coreceptor CCR5 or CXCR4 on the target-cell membrane, triggering fusion of the viral envelope with the plasma membrane. HIV can also bind to cell-surface lectins and enter cells by endocytosis, which is the predominant mode of entry into dendritic cells (DCs) and also occurs in macrophages. Once the viral core is released into the cytosol, HIV reverse transcriptase converts RNA into DNA in the reverse-transcription complex. That complex matures into the preintegration complex, which delivers reverse-transcribed HIV DNA to the nucleus for chromosomal integration. Few copies of HIV DNA integrate; thus, the bulk of HIV DNA is left behind in the cytosol to be cleared by host enzymes. Once the viral genomic DNA is integrated into a host chromosome, viral transcription is activated by host pathways with the assistance of the HIV protein Tat. HIV mRNAs are all capped and polyadenylated, like host RNA. The unspliced RNA is both translated to generate the Gag (group-associated antigen) and Pol (polymerase) proteins and incorporated as genomic RNA into nascent virions at cell-membrane sites where the envelope and capsid proteins assemble before budding (Fig. 2).
Mucosal innate immunity is the first line of defense against HIV-1 during the early phase of infection, and it also has a vital role in shaping ensuing adaptive immune responses. The types of innate immunity involved in HIV-1 infection can be divided into two major forms: cellular and intracellular. Cellular innate immunity includes functions of DCs, such as Langerhans cells, that are among the first group of cells that contact HIV-1 at the site of infection and that can mediate the trans-infection of CD4+ T cells49. The gd+ T cells offer innate responses to HIV by generating antiviral factors such as RANTES, MIP-1a and MIP-1b50. Natural killer (NK) cells also serve important roles in cellular innate immunity to HIV by eliminating infected cells and modulating DC functions51. Intracellular innate immunity includes intrinsic immunity mediated by host factors with important roles in restricting HIV-1 replication, such as APOBEC3G, TRIM5a, tetherin (BST-2) and SAMHD1 (refs. 52,53,54; Table 1). All of these restriction factors are also encoded by ISGs. HIV-1 counteracts some of these restriction factors via accessory proteins and avoids upregulation of other antiviral proteins encoded by ISGs in infected target cells55, 56.
Many steps of the HIV-1 life cycle are targeted by intrinsic antiviral factors such as TRIM5a, APOBEC3G, tetherin and SAMHD1. HIV-1 has evolved strategies to counteract these intrinsic antiviral factors, through accessory proteins such as Vif, Vpu and Vpx or other unknown mechanisms that are now under investigation. gRNA, genomic RNA; ssDNA, single-stranded DNA; dsDNA, double-stranded DNA.
APOBEC3
APOBEC3G (originally called CEM15) was one of the first intrinsic antiviral factors identified as acting against HIV-1. It was discovered by investigation of the function of the HIV-1 accessory protein Vif (viral infectivity factor)57, 58. Vif was known to be essential for HIV replication in certain cell lines (such as CEM-SS and SupT1 cells) but not in others (such as CEM or CD4+ T cells), and the effect of Vif was found to be dependent on virus-producing cells. Heterokaryon fusion of permissive and nonpermissive cells yielded cells with nonpermissive phenotypes, which suggested that a dominant antiviral factor exists in nonpermissive cells. This antiviral factor was identified as APOBEC3G through a cDNA expression screen for genes specifically expressed in nonpermissive cells and the ability to convert permissive cells into nonpermissive cells after expression57.
APOBEC3G belongs to a family of cytidine deaminases that includes seven members in primates (APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3DE, APOBEC3F, APOBEC3G and APOBEC3H). The mouse homolog of this family has only one member, encoded by Apobec3, which underscores the considerable evolutionary diversification that occurred at this locus. Indeed, the locus encoding APOBEC3 proteins shows strong evidence of positive selection during the evolution of primates59. APOBEC3G and APOBEC3F are the predominant restriction factors for HIV-1. They are packaged into HIV-1 virions through interaction with nucleocapsid portion of the HIV Gag protein. After infection of target cells and during reverse transcription, APOBEC3G edits C to U in single-stranded HIV DNA (negative strand), which results in G-to-A mutation in the HIV genome. G-to-A mutations often lead to premature stop codons that partially contributed to the diminished replication. Such G-to-A mutations are also frequently found in HIV DNA isolated from patients with AIDS60. APOBEC3G also inhibits reverse transcription and chromosomal integration through yet-to-be-defined mechanisms that are independent of its deaminase activity61, 62.
HIV-1 Vif counteracts APOBEC3G by promoting its ubiquitination by an E3 ligase complex consisting of Cul5, elongins B and C, and Rbx1. This ubiquitination targets APOBEC3G for degradation by the proteasome in virus-producing cells58. The interaction between Vif and APOBEC3G is species specific; for example, APOBEC3G from African green monkeys has a single-amino acid change at position 128 and is completely resistant to HIV-1 Vif-mediated degradation63, 64, 65, 66. Thus, this interaction is a major target for antiretroviral therapy at present.
TRIM5α
Restriction activity against HIV-1 in some nonpermissive cell lines can be saturated by a high multiplicity of infection67. One such restriction factor, originally called Ref1, restricts HIV-1 replication in lung fibroblasts from Old World monkeys. This restriction factor was subsequently determined to be TRIM5a, a member of the tripartite-motif (TRIM) family that shares a common organization at the amino terminus, which contains a RING domain, a B-box domain and a coiled-coil domain68. The RING domain is commonly found in E3 ubiquitin ligases, and the B-box domain determines substrate specificity. The carboxyl terminus of TRIM5a contains a B30.2 domain that binds to the capsid of the incoming virion and is most important for restriction. TRIM5a, specifically its B30.2 domain, is also a major determinant of the species tropism of retroviruses46. For example, human TRIM5a potently restricts murine leukemia virus (MLV) but does not restrict HIV-1 or simian immunodeficiency virus (SIV) from rhesus macaques (SIVmac). TRIM5a from rhesus macaques restricts HIV-1 but not SIVmac. The importance of TRIM5a and its capsid-binding activity was underscored again by the discovery of the TRIM5a-cyclophilin A fusion protein in cells from owl monkeys69. This protein is produced naturally by an in-frame fusion in which cyclophilin A replaces the B30.2 domain of TRIM5a. Cyclophilin A also binds to the HIV-1 capsid, and TRIM5a-cyclophilin A potently restricts HIV-1 through mechanisms similar to those used by TRIM5a.
TRIM5a blocks retrovirus replication early and before reverse transcription, probably during the process of uncoating46. As a ubiquitin E3 ligase, TRIM5a acts in both proteasome-dependent and proteasome-independent pathways. For example, inhibiting proteasomes during infection or disrupting the E3 ligase activity of TRIM5a only partially alleviates the restriction70, 71. TRIM5a promotes the rapid uncoating of HIV-1 capsids in vitro72. TRIM5a has also been found to promote innate immune signaling and to act as a PRR for the capsids of many retroviruses, including MLV, HIV and SIV73. TRIM5a expression in 293T human embryonic kidney cells activates AP-1 and NF-kB, but not type I interferons, by promoting the synthesis of free K63-linked polyubiquitin chains, which bind and activate the kinase TAK1.
Tetherin
Tetherin (also known as BST-2 or CD317) was discovered through the characterization of the HIV-1 accessory protein Vpu74, 75. Vpu enhances the release of HIV and other retroviral virions, thereby promoting replication. Similar to studies of Vif, heterokaryon fusion experiments suggested that a dominant restriction factor for Vpu-deficient HIV exists in nonpermissive cells. Such a factor was also found to be interferon inducible, as treatment with IFN-a converted permissive cells into cells that were nonpermissive to Vpu-deficient HIV76. Tetherin was later identified through comparative microarray analysis74. The topology of tetherin is unique and includes an amino-terminal cytoplasmic domain; a transmembrane domain; an extracellular, long, coiled-coil domain; and a carboxy-terminal glycosylphosphatidylinositol membrane anchor. The short cytoplasmic domain binds the clathrin adaptors for endocytosis. Tetherin is thought to hold virions at the cell surface by inserting the glycosylphosphatidylinositol membrane anchor into the virion envelope or by dimerization of two tetherin molecules, one anchored at the host cell membrane and one anchored at the virion envelope. Tethered virions are then internalized by endocytosis and are subsequently degraded in the endosomes77. Vpu promotes the degradation of tetherin, thereby facilitating HIV infection.
Tetherin targets many other enveloped viruses, such as other retroviruses (MLV and human T lymphotropic virus type 1), filoviruses (Ebola virus) and herpesvirus (Kaposi's sarcoma-associated herpesvirus)77. Each of these viruses counteracts tetherin via a viral protein that binds to tetherin and either promotes its degradation (the K5 protein of Kaposi's sarcoma-associated herpesvirus) or inhibits its function through an unknown mechanism (the glycoprotein of Ebola virus).
Like APOBEC3G and TRIM5a, tetherin also rapidly evolves under positive selection. Tetherin has species specificity that may have contributed to shaping the evolution of primate lentiviruses46. The genomes of many primate lentiviruses, mostly SIV isolates, do not encode Vpu. Such SIV isolates use negative factor (Nef) to counteract the simian orthologs of tetherin, which feature a five-amino acid insertion in the cytoplasmic domain that is not present in human tetherin. Nef specifically binds to the extra five amino acids in simian tetherin and is thus unable to target human tetherin. HIV-1 originated from the SIV of chimpanzees through a cross-species transmission to humans. The SIV of chimapanzees uses Nef to counteract chimpanzee tetherin, whereas HIV-1 has had to adapt and use Vpu to antagonize human tetherin to survive the new host environment (because the Nef-targeting motif is missing).
Tetherin also has a role in immune signaling. Tetherin is a ligand for ILT7, a membrane receptor selectively expressed in plasmacytoid dendritic cells. The binding of tetherin to ILT7 leads to the inhibition of TLR-mediated interferon responses in plasmacytoid DCs78. Tetherin has also been found to activate NF-kB in a large-scale screening79. The importance of this in viral infection remains to be shown, but a simple evasion strategy could be imagined in which enveloped viruses antagonize tetherin to promote virion release and at the same time downregulate NF-kB signaling to dampen the host immune response.
SAMHD1 and TREX1
HIV-1 replication is very inefficient in cells of the myeloid lineage, especially DCs. This myeloid-specific restriction can be overcome by treating cells with virus like particles containing the accessory protein Vpx from SIVmac or HIV-2 (ref. 80). The genome of HIV-1 does not encode Vpx, and Vpx-deficient SIVmac or HIV-2 fails to replicate in DCs. The host restriction factor targeted by Vpx has been identified as SAMHD1 (refs. 53,54). SAMHD1 seems to inhibit HIV-1 reverse transcription53 and innate immune responses to HIV (at least in monocyte-derived DCs)81. SAMHD1 is the only mammalian protein that contains both a SAM domain, predicted to mediate protein-protein interactions, and a HD domain, which has nucleotide-phosphohydrolase activity82. Vpx binds SAMHD1 and brings it to DCAF1 and DDB-CUL4 E3 ubiquitin ligase complexes for ubiquitination and subsequent degradation. Structural and biochemical studies of the HD domain have shown that SAMHD1 is a potent dGTP-stimulated triphosphohydrolase that converts deoxynucleoside triphosphates (dNTPs) to deoxynucleoside and triphosphate83. It has been shown that SAMHD1 regulates the dNTP pool in myeloid cells, thus blocking HIV reverse transcription by throttling the dNTP supply84. SAMHD1 may have an additional role in limiting innate immune signaling in response to HIV replication, particularly in DCs. Indeed, DCs rendered permissive to HIV-1 infection through the expression of Vpx, which causes SAMHD1 degradation, induce type I interferons through cellular IRF3 and cyclophilin A81. Thus, HIV-1 avoids infecting DCs so that it does not induce interferons but stealthily passes through DCs to facilitate its infection of helper T cells. Consistent with an inhibitory effect of SAMHD1 on interferon induction, mutations in SAMHD1 are associated with Aicardi-Goutieres Syndrome85, an autoimmune disorder and a neurological brain disease that is a genetic mimic of congenital viral infection. Patients with this syndrome have higher concentrations of IFN-a.
Interestingly, the product of TREX1, another gene associated with Aicardi-Goutieres syndrome, is also important for HIV replication, specifically by inhibiting innate immune responses to HIV DNA55. TREX1 is a 3' exonuclease that contains three well-conserved exonuclease motifs at its amino terminus and a hydrophobic region at the carboxyl terminus that is important for its localization to the cytoplasm and endoplasmic reticulum. In Trex1-/- mouse cells and in human CD4+ T cells and macrophages depleted of TREX1 by RNAi, cytosolic HIV DNA accumulates and HIV infection induces interferons that inhibit HIV replication and spreading. TREX1 binds to cytosolic HIV DNA and digests excess nonproductive HIV DNA that would otherwise activate interferon expression via a pathway dependent on the kinase TBK1, STING and IRF3 (ref. 55). TREX1 also prevents autoimmunity induced by DNA derived from endogenous retroelements86, which may explain the Aicardi-Goutieres syndrome in patients with loss-of-function mutations of TREX1.
Although both SAMHD1 and TREX1 are associated with the same autoimmune disease, they have opposite effects on HIV-1 replication. SAMHD1 is antiviral, whereas TREX1 is proviral, for HIV-1, through distinct mechanisms. Interestingly, both proteins seem to target the reverse-transcription step by limiting the dNTP supply (SAMHD1) or by inhibiting recognition by the immune system of nonproductive products of reverse transcription (TREX1). Reverse-transcribed HIV DNA can also trigger a proinflammatory response in nonproductively infected CD4+ T cells in the tonsils, which promotes T cell killing87. Reverse transcription of HIV RNA is becoming increasingly recognized as an important process in the HIV life cycle that is carefully regulated by the concerted effort of viral and host factors. Nucleic acids generated by reverse transcription are also targeted by host immunosurveillance. Further investigation is needed to provide additional insight into the dynamic interaction between the reverse transcription of HIV RNA and innate immune signaling and how this influences the establishment of infection and HIV pathogenesis.
Intrinsic immunity to influenza virus
Influenza A virus is a negative-sense single-stranded RNA virus of the orthomyxovirus family. Influenza A virus enters host cells through the attachment of viral hemagglutinin to host-cell receptors containing a-2,3- or a-2,6-linked sialic acid moieties, followed by endocytosis88 (Fig. 3). Acidification of the endocytosed vesicle promotes fusion of the viral envelope and the endosome membrane, followed by release of the viral RNA-protein complex into the cytoplasm. The viral RNA-protein complex then translocates into the nucleus, where negative-strand viral RNA is converted to complementary positive-strand RNA and mRNA. Viral mRNAs are exported to the cytoplasm for translation to generate a total of 11 viral proteins. Some of the viral proteins (M1 and NEP) are essential for genome replication and transcription, and they shuttle in and out of the nucleus to promote the production of more viral RNA-protein complexes. Other viral proteins are transported through the host protein-secretary pathway to the plasma membrane, where new viral particles form. The nonstructural protein NS1 inhibits host interferon-mediated antiviral responses and thus promotes the pathogenesis of influenza virus89.
The main target of influenza virus is epithelial cells in the respiratory tract. Macrophages and DCs in the airway can also be infected by influenza virus, and these cells have an important role in host innate and adaptive immune responses to the virus. Influenza virus can be recognized by many PRRs, including RLRs, TLRs and NLRs. In infected fibroblasts, the cytosolic RNA sensor RIG-I recognizes the 5' triphosphate of influenza virus genomic RNA and triggers interferon via MAVS and IRF3. In fact, studies of influenza virus have had an important role in elucidating the molecular mechanisms of the RIG-I signaling pathway25. As a counterstrike, the viral protein NS1 helps the virus to evade detection by the innate immune system by sequestering viral RNA or by binding to RIG-I and other proteins required for RIG-I signaling25. In plasmacytoid DCs, the single-stranded RNA genome of influenza virus can be recognized by TLR7 in the endosomes and induce the production of proinflammatory cytokines and interferons90. The TLR3-dependent inflammatory response has also been linked to influenza virus-infected lung epithelial cells and the induction of acute pneumonia91, 92. Infection with influenza virus also activates NLRP3 inflammasomes as an innate immune response that contributes to the adaptive immune response90. Initial evidence for NLRP3 activation was provided by the observation that influenza virus-infected human macrophages produce IL-1b and IL-18 through a caspase-1-dependent pathway93, but the mechanistic details began to emerge only recently. Influenza virus induces IL-1b production by enhancing the transcription of genes encoding pro-IL-1b and NLRP3 (signal 1) and by activating NLRP3 inflammasomes (signal 2)94, 95, 96. Signal 1 is triggered by the detection of viral RNA by TLR7, which activates NF-kB. Many sources contribute to signal 2, and all depend on the viral M2 protein, including ionic imbalance of the trans-Golgi pH, potassium efflux through the P2X7 receptor cation channel, and an increase in cellular reactive oxygen species. Studies of inflammasome-deficient mice have found that the inflammasome complex is dispensable for early clearance of the virus but is essential for late-stage clearance of the virus94, 95, 96. Further investigation is needed to elucidate the role of inflammasomes in mediating host innate and adaptive responses to influenza virus.
The IFITM family
The interferon-induced transmembrane genes belong to a family of small ISGs that includes IFITM1, IFITM2 and IFITM3. IFITM3 has been identified in two genome-wide screens (RNAi97 and yeast-two-hybrid98) as encoding a host restriction factor for influenza A virus. Expression of these genes is induced by infection with influenza virus. Knockdown via RNAi or deletion of these genes results in more influenza virus replication, and overexpression of IFITM proteins potently inhibits viral replication. IFITM proteins probably block infection early during entry97, although the mechanistic details remain unclear. Interestingly, avian cells do not seem to express a homolog to IFITM3, which raises the possibility that IFITM proteins might influence viral tropism. Moreover, IFITM proteins also inhibit the replication of some flaviviruses, including dengue virus and West Nile virus97. Infection with DNA viruses such as cytomegalovirus and herpes simplex virus also induces the expression of genes encoding IFITM proteins99, 100, although there is no clear evidence for an antiviral activity of IFITM proteins against DNA viruses.
IFITM proteins have also been linked to cancer101. IFITM1 and IFITM3 associate with the cell-surface antigen CD81, which is expressed in a variety of cancers. All IFITM proteins have a conserved CD225 domain with antiproliferative activity; such activity can be enhanced by interferon treatment102. Overexpression of IFITM3 inhibits the proliferation of interferon-sensitive melanoma cells, whereas knockdown of IFITM1 hampers IFN-g-mediated antiproliferative effects103. Comparison of genes encoding IFITM proteins of different organisms has shown higher than normal sequence variation, which suggests that these genes are under positive selection during evolution101. Together, the observations presented above indicate IFITM proteins represent likely targets for therapeutics against virus and cancer.
The IFIT family
The IFIT family (interferon-induced proteins with tetratricopeptide repeats) includes four members in humans: IFIT1 (ISG56), IFIT2 (ISG54), IFIT3 (ISG60) and IFIT5 (ISG58). All IFIT proteins are cytoplasmic proteins with multiple tetratricopeptide repeats, which are helix-turn-helix structures that mediate protein-protein interactions and the assembly of protein complexes104. IFIT1 was initially found to inhibit cellular translation by binding to the eIF3 initiation factor105. This activity is part of the nonspecific antiviral response triggered by interferons. IFIT1 also inhibits the cytoplasmic sensing of viral RNA by binding to STING, thereby disrupting the formation of signaling scaffolds106. This negative feedback of interferon signaling regulated by IFIT1 is considered important for the prevention of interferon overdrive in infected cells that may be toxic for the host.
IFIT proteins have been shown to recognize viral RNA that contains a 5'-triphosphate moiety or lacks 2'-O-methylation107, 108, 109. Cellular mRNA usually contains a 5' guanosine cap that stabilizes the mRNA for translation and differentiates self from non-self viral genomic RNAs that often contain 5' triphosphate. Cellular mRNAs are also methylated at the 2'-O position, but the purpose of this has been unclear because it does not contribute to translation or stability. The genomes of many RNA viruses also encode a methyltransferase that methylates the 2'-O position of viral RNA to mimic host mRNA. It is now clear that this modification is important for these viruses to evade host restriction by IFIT proteins. Viruses defective in methyltransferase show enhanced sensitivity to interferon treatment in an IFIT-dependent manner107, 108. Thus, 2'-O-methylation of host mRNAs is critical for the distinction between self and non-self, for at least some RNA viruses. Interestingly, the mRNA of plants or plant viruses does not contain 2'-O-methylation and does not have interferon responses or orthologs of genes encoding IFIT proteins. This represents yet another evolutionary sophistication acquired by vertebrates. IFIT1 also binds viral genomic RNA containing 5' triphosphate, similar to RIG-I, and exerts a direct antiviral activity109. Instead of activating interferons, the binding of IFIT proteins to 5'-triphosphate viral RNA inhibits viral translation and replication. Consistent with the direct antiviral activity of IFIT1, knockdown of IFIT1 results in more viral replication without affecting the interferon response. Moreover, this antiviral activity is orchestrated by at least three members of the IFIT family as a protein complex. The structural basis for IFIT recognition of 5' triphosphate and the absence of 2'-O-methylation in RNA molecules is unclear. Similarly, the mechanisms of inhibition after recognition by IFIT proteins remain to be determined.
MxA
Human MxA is a GTPase with broad antiviral activities. MxA has domain structures similar to those of the dynamin family of large GTPases, including an amino-terminal GTPase domain, a self-oligomerization domain and a carboxy-terminal GTPase effector domain. Both human MxA and mouse Mx1 have antiviral activity against influenza virus infection. Mx1-deficient mice succumb to infection with influenza virus, whereas wild type mice are very resistant and survive high-dose challenges110, 111. Mouse Mx1 localizes to the nucleus and blocks primary transcription of influenza virus RNA. Human MxA is cytoplasmic and blocks the late life cycle of influenza virus, such as secondary transcription and viral replication112. Different strains of influenza virus vary in their sensitivity to these proteins, which is influenced by the viral nucleoprotein113, 114. The structure of MxA has shown that it might form an oligomeric ring structure around viral nucleocapsid, thereby inhibiting viral replication115. It is unclear how human MxA can inhibit a broad spectrum of RNA viruses, some of which replicate in the nucleus, and whether MxA acts by recognizing a common viral component or structure.
Conclusions and perspectives
The rapid progress in innate immunity research in the past decade has been breathtaking. Several families of innate immune sensors have been discovered and the signaling pathways that they trigger are being rigorously investigated. By comparison, less is understood of the intrinsic antiviral factors and their mechanisms of action. The intense medical interest in HIV, influenza virus and other viruses has begun to elucidate some of the host restriction factors that limit infection by these viruses. Such studies have also provided striking examples of the 'arms race' between the host and virus during their coevolution. Further studies of individual viral proteins will continue to shed light on the host immunity that attacks various viruses. Conversely, investigation of host antiviral factors will elucidate how viruses evade the host's immunosurveillance. Studies of viral tropism in certain cell types have had an excellent track record of discovering important antiviral factors, such as APOBEC3G, TRIM5a, tetherin and SAMHD1. There is no reason to think that such a successful track record will end any time soon.
Technological advances such as genome-wide RNAi screens have provided additional avenues for discovering previously unknown host antiviral factors, as exemplified by the success in identifying many host proteins and noncoding RNAs (such as microRNA) that either permit or impede the replication of HIV and influenza virus. As experience is gained and new tools are developed for differentiating true positive results from false-negative or false-positive results, large-scale screening approaches will be applied to more viruses with better success rates. The next challenge will be to investigate the function of newly identified antiviral factors and elucidate their mechanisms of action. Knowledge gained from such studies will be very powerful for the design of the future generation of antiviral therapies.
The repeated failure to produce an effective HIV vaccine is a humbling reminder that full understanding of the immune response to HIV is still lacking. In fact, very little is known about innate immune responses to retroviruses in general. Even the existence of intracellular innate immunity to retroviruses is a subject of debate, because retroviral infection normally does not trigger the production of cytokines or interferons. However, retroviruses, including HIV, clearly activate T cells and B cells in vivo. A study has shown that TLR7 detects entry of the mouse retrovirus MMTV into host cells and activates humoral immune responses116. Emerging literature has also shown the importance of natural killer cells as the cytotoxic arm of innate immunity during HIV transmission51, 117. The findings that HIV ceases replication in 'professional' interferon-producing cells such as DCs because of the presence of SAMHD1 and that HIV coopts the host protein TREX1 to suppress interferon production in target cells have reinvigorated research into innate and intrinsic immunity to HIV. This line of research may be key to the development of effective treatments for HIV and other devastating pathogens.
| |
| |
| |
|
|
|