| |
Sustained Efficacy and Safety of Raltegravir After 5 Years of Combination Antiretroviral Therapy as Initial Treatment of HIV-1 Infection: Final Results of a Randomized, Controlled, Phase II Study (Protocol 004)
|
| |
| |
Download the PDF here
JAIDS Journal of Acquired Immune Deficiency Syndromes: 1 September 2012
"This phase II clinical trial began as a 48-week dose-ranging study of raltegravir versus efavirenz, both in combination with tenofovir/lamivudine, as initial therapy of HIV-1 infection. At week 48, all of the raltegravir doses studied (100, 200, 400, and 600 mg bid) compared favorably with efavirenz on both efficacy and safety parameters, supporting the consolidation of all raltegravir dosing arms to receive 400 mg bid.4 The study was extended to 240 weeks, providing the longest duration of raltegravir therapy in treatment-naive patients to date. The final 5-year study results presented in this report demonstrate a sustained profile of both durable efficacy and favorable safety."
"Raltegravir remained generally well tolerated during long-term use, with few discontinuations and no drug-related serious adverse events. In addition, raltegravir maintained a neutral effect on fasting lipids (LDL-cholesterol, total cholesterol, and triglycerides). These efficacy and safety data are consistent with available data from STARTMRK, an ongoing phase III study of raltegravir versus efavirenz, both in combination with tenofovir/emtricitabine.6-8 The potent efficacy and favorable safety profile confirm that raltegravir, in a combination antiretroviral regimen, is an appropriate treatment option for initial therapy of HIV infection"
"Virologic failure occurred by year 5 in 10 (6%) of 160 patients in the raltegravir group and 5 (13%) of 38 patients in the efavirenz group.....
Serious adverse events and discontinuations due to adverse events were infrequent in both groups, and few drug-related adverse events were reported after week 48. Neurotoxicities, such as headache, dizziness, and sleep disorders were observed more frequently in the efavirenz group than in the raltegravir group. elevated CPK was more common in the raltegravir group than in the comparator group but was not associated with clinical manifestations and did not lead to treatment discontinuation; these observations are consistent with the phase III BENCHMRK studies in treatment-experienced patients.13 Dyslipidemia is a common complication of current antiretroviral therapies19-21 and is typically associated with protease inhibitors and certain nucleoside reverse transcriptase inhibitors. In the present study, serial measurement of fasting lipids over 240 weeks revealed no adverse impact of raltegravir on serum lipids.. At week 240, mean elevations of high-density lipoprotein (HDL)-cholesterol and total cholesterol were smaller in the raltegravir group than in the efavirenz group (7.4 vs 14.2 mg/dL, P = 0.024; 11.7 vs 26.4 mg/dL, P = 0.014), while the mean changes in low-density lipoprotein (LDL)-cholesterol and triglycerides were not significantly different between treatment groups. The mean change in the ratio of total to HDL-cholesterol was -0.5 for the raltegravir group and -0.6 for the efavirenz group (from 4.6 at baseline in both groups). Lipid-lowering medication was taken by 3.8% of raltegravir recipients and 2.6% of efavirenz recipients prior to study entry and by 10.6% and 13.2%, respectively, during the study."
Gotuzzo, Eduardo MD*; Markowitz, Martin MD; Ratanasuwan, Winai MD; Smith, Graham MD; Prada, Guillermo MD; Morales-Ramirez, Javier O. MD; Strohmaier, Kim M. BS#; Lu, Chengxing PhD; Bhanja, Sanhita MS**; Nguyen, Bach-Yen MD; Teppler, Hedy MD; for the Protocol 004 Study Team
Abstract
Abstract: Raltegravir as initial HIV therapy was examined in a double-blind study; 160 patients were randomized to raltegravir (400 mg bid after dose-ranging), 38 to efavirenz, both with tenofovir/lamivudine. At week 240, HIV-RNA remained <50 copies per milliliter in 68.8% (raltegravir) versus 63.2% (efavirenz), and CD4 increases were 302 versus 276 cells per microliter, respectively. Early HIV-RNA decline predicted later CD4 increases in both groups. Raltegravir resistance was observed in 3 of 10 raltegravir recipients with virologic failure. Few drug-related adverse events were reported after week 48. Raltegravir had minimal effect on laboratory values, including lipids. Raltegravir with tenofovir/lamivudine showed durable efficacy and good tolerability over 5 years.
INTRODUCTION
Raltegravir is a strand-transfer inhibitor of HIV-1 integrase1 that is approved for use in combination with other antiretroviral agents for the treatment of HIV-1 infection.2 Raltegravir has shown potent efficacy and good tolerability in treatment-naive patients3-8 and in treatment-experienced patients with virologic failure and multidrug-resistant HIV-1.9-13 Long-term efficacy and safety data are essential to distinguish among antiretroviral regimens because treatment of HIV infection is generally life-long, and many patients have or will develop comorbid conditions that could influence the choice of antiviral therapies. The first clinical trial of raltegravir as initial therapy in HIV-1 infection (Protocol 004) was extended to 240 weeks (5 years). This report presents the final 5-year efficacy and safety data from this study, along with an exploratory analysis of the relationship between early virologic response and long-term change in CD4 counts.
METHODS
Protocol 004 (NCT # 00100048) was a double-blind, randomized, dose-ranging study in treatment-naive HIV-1-infected patients conducted at 29 sites in the United States, Canada, Latin America, Thailand, and Australia from June 14, 2005 to July 12, 2010. Eligibility criteria included: HIV-1 RNA ≥5000 copies per milliliter, CD4+ T-cells ≥100 per microliter, and documented genotypic and phenotypic susceptibility to efavirenz, tenofovir, and lamivudine. Part I consisted of 10 days of raltegravir monotherapy versus placebo in 35 patients.3 Part II examined the safety, tolerability, and efficacy of raltegravir 100, 200, 400, or 600 mg twice daily versus efavirenz 600 mg per day, each given with tenofovir 300 mg per day and lamivudine 300 mg per day, for 48 weeks in 30 patients continued from Part I (Cohort I) and 171 patients randomized into Part II (Cohort II).4 No difference in efficacy or tolerability was observed among the raltegravir doses studied at either 10 days or 48 weeks. After 48 weeks, the protocol entered a double-blind extension, which consolidated all raltegravir groups to receive 400 mg bid; all other study medications were continued unchanged. Physical examinations, laboratory tests, and virologic and immunologic responses were assessed every 12 weeks. In this report, all results are shown with the initial raltegravir dose groups combined, to compare with the efavirenz group. Details of the study design and methods, and results for the initial raltegravir dose groups have been previously reported.4
Exploratory Analysis
Earlier HIV suppression by raltegravir compared with efavirenz was observed in this study4 and in the STARTMRK study.6 To better understand the impact of this early suppression on long-term efficacy outcomes, the relationship between early decrease in HIV RNA and later CD4-cell count was explored using the observed-failure approach. Separate linear regression models of CD4 cell count were created at each yearly time point through year 5 (week 240), with the following variables among model predictors: baseline CD4 cell count, decrease in log HIV RNA at week 8, and treatment group. The significance threshold was set at P > 0.05.
RESULTS
Baseline Patient Characteristics
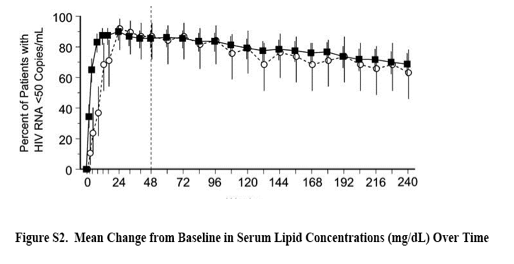
A total of 198 patients received study therapy in the combination phase of this protocol. Baseline characteristics were balanced between the aggregate raltegravir and efavirenz groups (see Table S1, Supplemental Digital Content, http://links.lww.com/QAI/A339). Plasma HIV-1 RNA was >100,000 copies per milliliter in 34% of patients overall. Mean HIV-1 RNA levels were 4.7 and 4.8 log10 copies per milliliter for the raltegravir and efavirenz groups, respectively, and mean CD4+ T-cell counts were 305 and 280 cells per microliter, respectively. A total of 142 patients completed 240 weeks of study therapy, 116 (72%) of the raltegravir group and 26 (67%) of the efavirenz group; specific reasons for study discontinuation are provided in the Supplemental Digital Content (see Figure S1, http://links.lww.com/QAI/A339).
Efficacy
Over 5 years of treatment, raltegravir displayed potent efficacy comparable to that of efavirenz on both virologic and immunologic measures (Figs. 1A-C). HIV-1 RNA remained suppressed to <50 copies per milliliter in 68.8% of patients in the raltegravir group and 63.2% in the efavirenz group. Efficacy outcomes were comparable between patients with high baseline HIV-1 RNA (>100,000 copies per milliliter) and those with baseline viral load ≤100,000 copies per milliliter (see Table S2, Supplemental Digital Content, http://links.lww.com/QAI/A339). In the exploratory analysis, baseline CD4 count and early (week 8) decline in log HIV RNA were significant predictors for CD4 count at each yearly time point (all P < 0.001). Treatment effect was not a significant predictor in the exploratory models, indicating that with the same level of baseline CD4 count and early decline in log HIV RNA, the CD4 cell counts were not significantly different between treatment groups at each yearly time point.
Virologic failure occurred by year 5 in 10 (6%) of 160 patients in the raltegravir group and 5 (13%) of 38 patients in the efavirenz group. Integrase genotype data were available for 8 patients who experienced virologic failure while receiving raltegravir and had sufficient virus for amplification. Signature integrase resistance mutations were demonstrated in 3 of these patients, including N155H (2 patients) and Y143C (1 patient); these 3 patients also displayed resistance to lamivudine, and one also showed resistance to tenofovir. Of the remaining 5 patients, one was resistant to lamivudine only, and 4 had no evidence of resistance to any drug in the regimen. Among the 5 patients with virologic failure on efavirenz, 2 had evidence of resistance to efavirenz, 1 showed resistance to tenofovir/lamivudine, and 2 showed no resistance to any drug in the regimen. Specific treatment-emergent mutations are listed in the Supplemental Digital Content (see Table S3, http://links.lww.com/QAI/A339). Phenotypic data were generally consistent, with the exception of one patient who showed reduced phenotypic susceptibility to efavirenz in the absence of resistance-associated mutations (data not shown).
Safety
Frequencies of clinical and laboratory adverse events are shown in Table 1 for the following time intervals: weeks 0-48, weeks 48-240, and weeks 0-240. Few drug-related clinical adverse events were reported after week 48 in either treatment group (raltegravir 7.5%; efavirenz 5.3%), and the most common drug-related adverse events after 5 years were essentially the same as those observed after 1 year. Neuropsychiatric adverse events occurred in 25.0% of the raltegravir group and 34.2% of the efavirenz group after week 48. The difference between treatment groups in neuropsychiatric events at week 240 [38.1% vs 63.2%; 95% confidence interval (CI): -40.7 to -7.4] was mainly due to dizziness, abnormal dreams, and nightmares; depression-related events occurred with similar frequency in both groups (raltegravir 15.0%, efavirenz 13.2%; 95% CI: -13.1 to 11.9).
By week 240, serious clinical adverse events had occurred in 15.6% of raltegravir recipients and 10.5% of efavirenz recipients. In the raltegravir group, one serious clinical adverse event (osteonecrosis) was considered by the investigator to be possibly related to tenofovir; this patient remained on the full-study regimen. In the efavirenz group, one serious clinical adverse event (gastrointestinal carcinoma) was considered drug-related and resulted in discontinuation from the study. There were no serious laboratory adverse events in either treatment group.
Grades 3/4 laboratory abnormalities were uncommon during this 5-year study (Table 1). The most common abnormality in the raltegravir group was elevated creatine phosphokinase (CPK), occurring in 15 patients (9.4%) versus 2 (5.4%) in the efavirenz group. Eleven of the 15 cases in the raltegravir group occurred after week 48, and 8 cases were considered related to strenuous exercise. No cases were associated with clinical myopathy, myositis, or rhabdomyolysis. Raltegravir was temporarily interrupted in one case, a grade 4 CPK elevation that was considered possibly drug related, which did not recur upon rechallenge. All CPK elevations were transient and resolved; none required permanent discontinuation of study therapy.
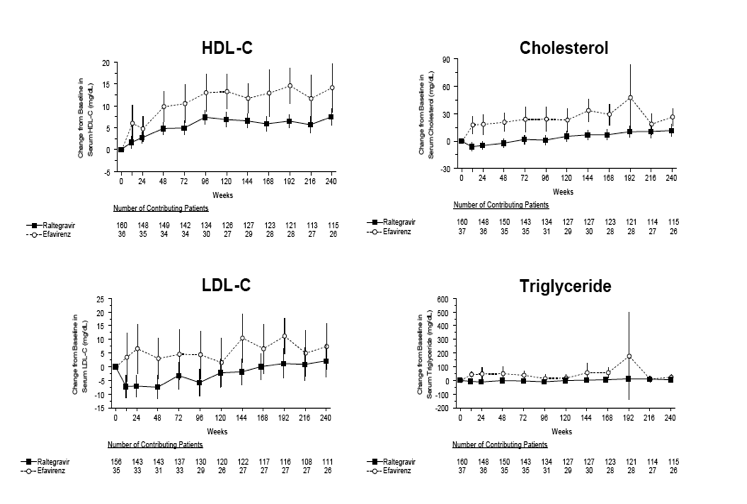
The mean changes from baseline in fasting lipid levels over the entire study period are shown in the Supplemental Digital Content (see Figure S2, http://links.lww.com/QAI/A339). At week 240, mean elevations of high-density lipoprotein (HDL)-cholesterol and total cholesterol were smaller in the raltegravir group than in the efavirenz group (7.4 vs 14.2 mg/dL, P = 0.024; 11.7 vs 26.4 mg/dL, P = 0.014), while the mean changes in low-density lipoprotein (LDL)-cholesterol and triglycerides were not significantly different between treatment groups. The mean change in the ratio of total to HDL-cholesterol was -0.5 for the raltegravir group and -0.6 for the efavirenz group (from 4.6 at baseline in both groups). Lipid-lowering medication was taken by 3.8% of raltegravir recipients and 2.6% of efavirenz recipients prior to study entry and by 10.6% and 13.2%, respectively, during the study.
DISCUSSION
This phase II clinical trial began as a 48-week dose-ranging study of raltegravir versus efavirenz, both in combination with tenofovir/lamivudine, as initial therapy of HIV-1 infection. At week 48, all of the raltegravir doses studied (100, 200, 400, and 600 mg bid) compared favorably with efavirenz on both efficacy and safety parameters, supporting the consolidation of all raltegravir dosing arms to receive 400 mg bid.4 The study was extended to 240 weeks, providing the longest duration of raltegravir therapy in treatment-naive patients to date. The final 5-year study results presented in this report demonstrate a sustained profile of both durable efficacy and favorable safety.
In the week 240 analysis of efficacy, raltegravir demonstrated sustained virologic and immunologic responses, as compared with efavirenz. Similar proportions of patients maintained HIV RNA <50 copies per milliliter (68.8% for raltegravir vs 63.2% for efavirenz), consistent with previous analyses at weeks 48, 96, 144, and 192.4,5,14,15 CD4 T-cell counts showed continued increases, with gains of 302 cells per microliter for raltegravir and 276 cells per microliter for efavirenz at week 240. In an exploratory analysis, early decline in log HIV RNA was a significant predictor of later increases in CD4 T-cell count regardless of treatment group. Virologic failure was infrequent in both treatment groups. Resistance to raltegravir was observed in 3 of the 10 patients who experienced virologic failure on raltegravir, suggesting that there were reasons for failure other than development of raltegravir resistance. The observed raltegravir resistance mutations were consistent with those observed in other clinical studies of raltegravir.8,10,13
In the week 240 final safety analysis, no clinically meaningful differences were observed in the overall adverse experience profile of raltegravir as compared with earlier analyses. Serious adverse events and discontinuations due to adverse events were infrequent in both groups, and few drug-related adverse events were reported after week 48. Neurotoxicities, such as headache, dizziness, and sleep disorders were observed more frequently in the efavirenz group than in the raltegravir group. Four cases of exacerbation of existing depression associated with raltegravir have been reported in the literature.16 In the current study and in the phase III studies of raltegravir, depression occurred at generally similar rates between raltegravir and the comparator agent.17
Laboratory adverse events and grade 3 or 4 laboratory abnormalities of aspartate aminotransferase and alanine aminotransferase were infrequent and occurred at similarly low frequencies in both groups. Four cases of rhabdomyolysis temporally associated with raltegravir use have been reported in the literature.18 In the current study, elevated CPK was more common in the raltegravir group than in the comparator group but was not associated with clinical manifestations and did not lead to treatment discontinuation; these observations are consistent with the phase III BENCHMRK studies in treatment-experienced patients.13 Dyslipidemia is a common complication of current antiretroviral therapies19-21 and is typically associated with protease inhibitors and certain nucleoside reverse transcriptase inhibitors. In the present study, serial measurement of fasting lipids over 240 weeks revealed no adverse impact of raltegravir on serum lipids.
In treatment-naive HIV-infected patients, raltegravir 400 mg bid, in combination with tenofovir and lamivudine, demonstrated potent and durable efficacy over 5 years of treatment, with sustained virologic suppression below 50 copies per milliliter and continued increases in CD4 counts. Raltegravir remained generally well tolerated during long-term use, with few discontinuations and no drug-related serious adverse events. In addition, raltegravir maintained a neutral effect on fasting lipids (LDL-cholesterol, total cholesterol, and triglycerides). These efficacy and safety data are consistent with available data from STARTMRK, an ongoing phase III study of raltegravir versus efavirenz, both in combination with tenofovir/emtricitabine.6-8 The potent efficacy and favorable safety profile confirm that raltegravir, in a combination antiretroviral regimen, is an appropriate treatment option for initial therapy of HIV infection.
| |
| |
| |
|
|
|